(Cu,N)共摻雜TiO2/MoS2異質結的電子和光學性能:雜化泛函HSE06?
2018-12-14 03:01:56王冠仕林彥明趙亞麗姜振益張曉東
物理學報 2018年23期
關鍵詞:體系
王冠仕 林彥明 趙亞麗 姜振益 張曉東
(西北大學現代物理研究所,陜西省理論物理前沿重點實驗室,西安 710069)
(2018年8月12日收到;2018年9月29日收到修改稿)
1 引 言
在過去的幾十年中,研究者已經嘗試了大量的策略來解決以上問題.其中,常用方法是在光催化材料中摻雜各種雜質原子來調節材料的帶隙,例如,非金屬C,N和B摻雜TiO2[16?18],金屬Fe和Cu摻雜TiO2[19,20],能夠將光吸收邊緣擴展到可見光范圍并提高光催化效率.然而,單原子摻雜依舊存在一些缺陷,例如熱穩定性低、電子-空穴復合高等.在此基礎上,為了有效提高材料的光催化性能,對共摻雜體系進行研究,例如,非金屬-非金屬共摻雜、金屬-金屬共摻雜和金屬-非金屬共摻雜[21?23].此外,構建半導體異質結構是一種通過分離與轉移光生電子和空穴來提高半導體材料的光催化活性的有效方法.例如,研究發現TiO2/g-C3N4異質結構能夠有效分離光生電子和空穴,從而增強光催化產氫性能[24],SrTiO3/TiO2異質結有利于光生電子和空穴的快速分離,實現光催化性能的提高.因此,選擇合適的共摻雜雜質原子以及構建合適的半導體異質結能夠抑制光生電子-空穴對的復合和增強光催化活性.
最近,Jaiswal等[25]成功制備了(Cu,N)共摻雜TiO2材料,實驗結果表明,(Cu,N)共摻雜TiO2可以增強對可見光的吸收同時降低光生電子空穴的復合.在光催化制氫的研究中,人們發現材料表面與水接觸具有良好的光催化能力,而在自然界中,銳鈦礦型TiO2的晶面暴露在真空外的僅有(101)和(001)兩個表面,其中TiO2(101)表面占其中的94%,性能最穩定.現在有很多研究者對銳鈦礦型TiO2(101)表面進行了理論研究,例如Sun等[26]和Yang等[27]研究的銳鈦礦型TiO2(101)表面的光催化性.然而,據我們所知,關于(Cu,N)共摻雜的銳鈦礦型TiO2(101)表面的電子結構與光學性質方面的理論研究很少.因此,本文通過理論計算分析共摻雜對TiO2(101)表面光吸收的影響.另外,已有研究表明構建TiO2/MoS2異質結可以有效抑制載流子復合,從而提高TiO2材料的光催化活性[28?33].因此,根據上述分析,可以預測(Cu,N)共摻雜的TiO2/MoS2異質結可以有效地分離和轉移光生電子與空穴,同時增加對可見光的吸收.
在本文中,我們采用雜化泛函Heyd-Scuseria-Ernzerhof(HSE06)的方法系統地計算了Cu/N(共)摻雜的銳鈦礦型TiO2(101)表面和Cu/N(共)摻雜的TiO2/MoS2異質結的幾何結構、電子結構和光學性質.對于Cu/N(共)摻雜的銳鈦礦型TiO2(101)表面和TiO2/MoS2異質結,分別計算了Cu/N原子摻雜在不同的間隙和替位摻雜位置的性質.另外,我們還分析了Cu/N(共)摻雜的TiO2/MoS2異質結界面處的電荷轉移,研究了壓力對TiO2/MoS2異質結電子結構和光吸收性質的影響.本文試圖通過構建異質結、對異質結進行摻雜和加壓來提高TiO2材料的光催化活性.
2 計算模型和方法
本文構建的基礎模型有TiO2(101)表面和單層MoS2.為保證構建模型的穩定性,我們對其真空層進行測試,測試結果如圖1所示.
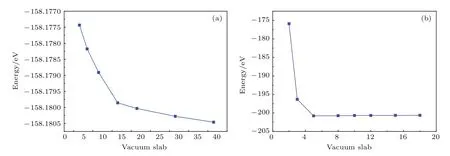
圖1 不同真空層的(a)TiO2(101)表面和(b)單層MoS2的總能量Fig.1 .Calculated total energy for(a)TiO2(101)surface and(b)MoS2monolayer with dif f erent vacuum spaces.

圖2 優化后的(a)銳鈦礦型TiO2(101)表面,(b)單層MoS2和(c)TiO2/MoS2異質結Fig.2 .Optimized structures:(a)TiO2(101)surface;(b)MoS2monolayer;(c)TiO2/MoS2heterojunction.
如圖1所示,TiO2(101)表面體系的總能量在真空層大于15 ?之后趨于穩定,因此本文在構建TiO2(101)表面時設置真空層為15 ?;單層MoS2體系的總能量當真空層大于6 ?后趨于穩定,因此單層MoS2的真空層被設為6 ?.銳鈦礦型TiO2(101)表面包含24個氧原子和12個鈦原子(共36個原子),優化后的結構如圖2(a)所示.單層MoS2包含8個硫原子和4個鉬原子(共12個原子),結構優化后如圖2(b)所示.利用優化好的TiO2(101)表面和單層MoS2結構構建的TiO2/MoS2異質結如圖2(c)所示.為得到穩定的TiO2/MoS2異質結,采用的真空層為15 ?,整個超胞的高度為35 ?.在研究摻雜對體系的影響時,考慮了Cu/N(共)摻雜TiO2(101)表面和TiO2/MoS2異質結的各種摻雜位置,例如N替位O(N@O)或N替位Ti(N@Ti),Cu替位O(Cu@O)或Cu替位Ti(Cu@Ti)等.
本文所有計算都使用VASP(Vienna ab initio simulation package)軟件包采用PAW 方法進行[34],利用廣義梯度中的Perdew-Burke-Ernzerhof(PBE)贗勢來描述其交換關聯和相關勢[35].其截斷能設置為400 eV,TiO2(101)表面和TiO2/MoS2異質結的幾何優化和電子性質計算中k點網格分別采用5×5×1和3×3×1.優化結構設置能量和力的精度分別為10?5eV和0.01 eV/?,為了獲得更為準確的優化結果,采用了DFT-D2方法對范德瓦耳斯(van der Waals,VDW)相互作用進行理論修正,并用優化好的結構計算其電子和光學性質.為了準確計算材料的電子和光學性質,采用雜化泛函(HSE06)方法,交換關聯能的表達公式如下:
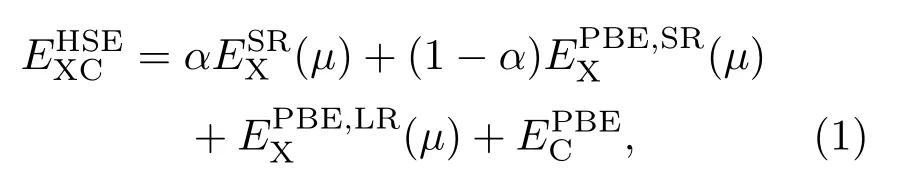
式中下標X和C分別表示交換泛函和關聯泛函;μ為篩選參數,設置為0.20 ??1;電子-電子相互作用可分為短程(shot range,SR)和長程(long range,LR)兩部分,通過混合參數α來確定,α的默認設置為0.20,但不同材料α值不同.為了準確計算出材料的電子和光學性質,通過計算材料帶隙測試了參數α的取值,結果如圖3所示.由圖3可以發現,α分別取0.05和0.17時單層MoS2和TiO2(101)表面的帶隙與實驗值(1.90 eV,2.90 eV)符合,在測出單層MoS2和TiO2(101)表面α值的基礎上,通過對兩個結構α值做加權平均,計算出TiO2/MoS2異質結的α值為0.14.
漢麻籽油的皂化、甲酯化根據GB/T 17376-2008/ISO 5509:2000“動植物油脂 脂肪酸甲酯制備”[11],選用三氟化硼法,每個樣品皂化、甲酯化3次。
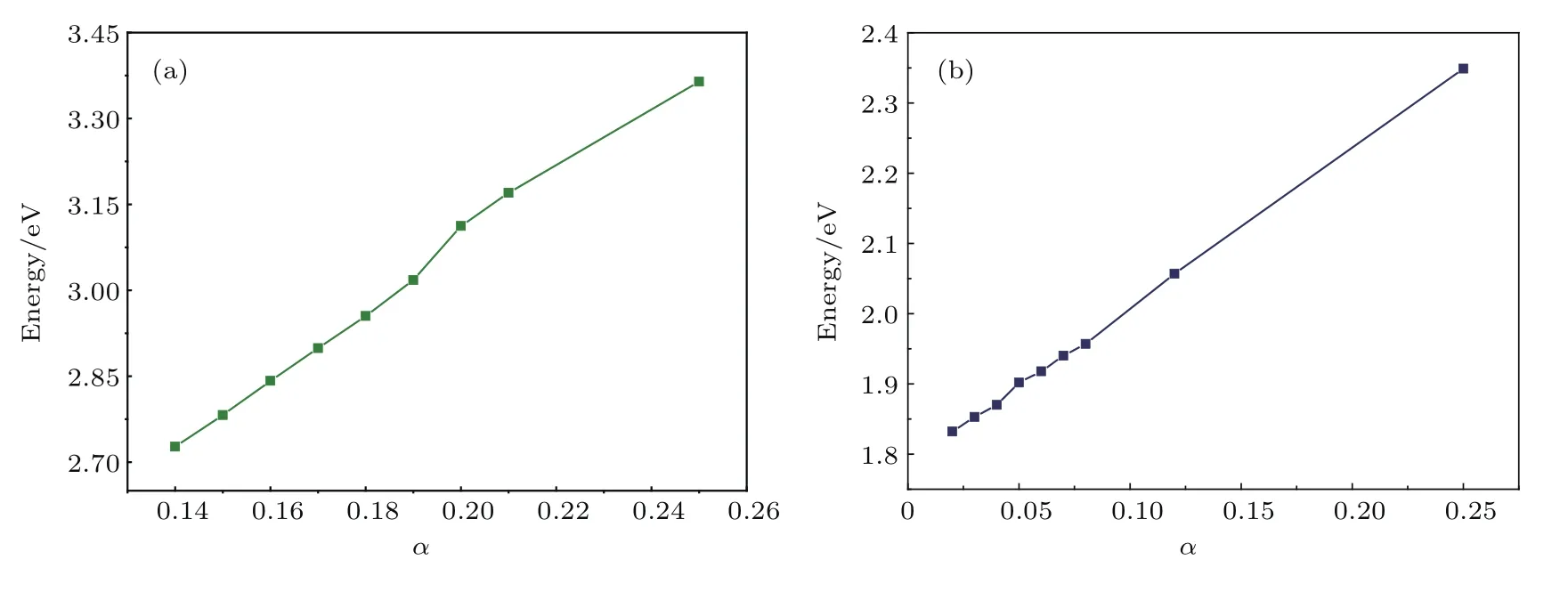
圖3 α參數為不同值時(a)銳鈦礦TiO2(101)表面和(b)單層MoS2的帶隙Fig.3 .Calculated band gap with dif f erent α parameters:(a)TiO2(101)surface;(b)MoS2monolayer.
3 結果與討論
3.1 幾何結構
在研究摻雜體系和復合體系的性質前,首先分別研究了銳鈦礦型TiO2(101)表面和單層MoS2結構的性質.鈦礦型TiO2(101)表面優化后,晶格常數為a=b=3.786 ?,幾何結構呈鋸齒狀,MoS2優化后的晶格常數a=b=3.19 ?,與以前的實驗數據相符合[36?39],這表明我們的計算方法是正確的.
優化后的Cu/N(共)摻雜的TiO2(101)表面的幾何結構如圖4所示,相應體系的平均鍵長如表1所列.銳鈦礦型TiO2(101)表面中Ti—O鍵的平均鍵長為2.001 ?,N摻雜TiO2(101)表面后,結構中Ti—O鍵的平均鍵長變化不明顯,然而Cu摻雜對Ti—O鍵的影響相對較大,可能是由于Cu原子摻雜引起了局域晶格畸變.N原子摻雜體系中,Ti—N鍵的平均鍵長與Ti—O鍵的平均鍵長近似相等,這可能是由于N原子的半徑(0.75 ?)與O原子(0.65 ?)的半徑相差較小.由于Cu原子的半徑(1.57 ?)遠遠大于O原子的半徑(0.65 ?),所以Cu摻雜體系和(Cu,N)共摻雜體系中Cu—Ti的平均鍵長(2.479 ?,2.627 ?)遠遠大于純的TiO2(101)表面中Ti—O的平均鍵長.這些結果表明,(Cu,N)共摻雜可以引起銳鈦礦型TiO2(101)表面晶格發生明顯的畸變,進而改變偶極矩,使得光生電子和空穴對更容易分離.在單層MoS2中,Mo—S的鍵長為2.411 ?,整個結構呈現為三明治形狀,與實驗結果相符合[40].
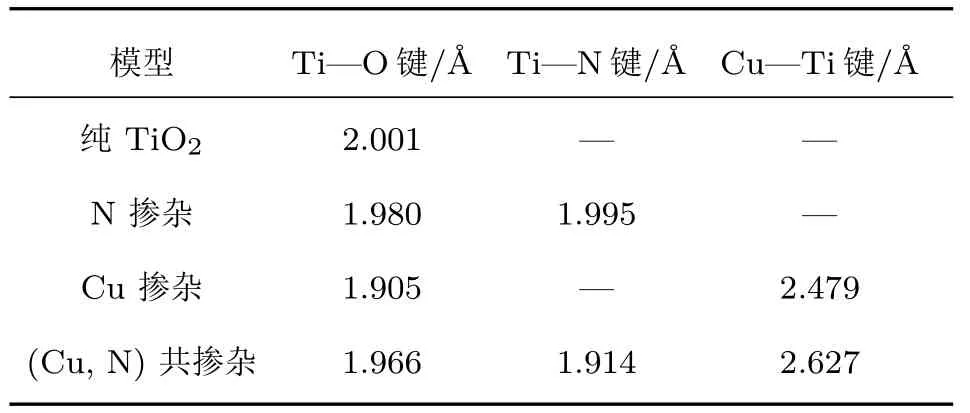
表1 優化后的Cu/N(共)摻雜的銳鈦礦型TiO2(101)表面的平均鍵長(單位:?)Table 1 .Average bond lengths of Cu or/and N doped anatase TiO2(101)surface after geometry optimization(unit:?).
把優化好的Cu/N(共)摻雜TiO2(101)表面和單層MoS2構建出TiO2/MoS2異質結,再次對TiO2/MoS2異質結進行幾何優化,其單層MoS2的厚度(h)和Cu/N(共)摻雜TiO2(101)表面與單層MoS2之間的垂直距離(D)的結果如表2所列.在純的TiO2/MoS2異質結中,單層MoS2的厚度和TiO2(101)表面與單層MoS2之間的垂直距離分別為h=2.964 ?和D=2.891 ?.當摻雜N原子后,MoS2厚度(h)和TiO2(101)表面與單層MoS2之間垂直距離(D)變化不大.Cu摻雜和(Cu,N)共摻雜TiO2/MoS2異質結中單層MoS2的厚度(h)均有所增大,同時Cu摻雜和(Cu,N)共摻雜體系的TiO2(101)表面與單層MoS2之間的垂直距離(D)相比于純的TiO2/MoS2異質結明顯減小,這是由于較長的Cu—Ti鍵減小了Cu原子與單層MoS2之間的垂直距離,從而增強了TiO2(101)表面與單層MoS2之間的相互作用力.這些異質結中的垂直距離(D)均為范德瓦耳斯平衡間距.
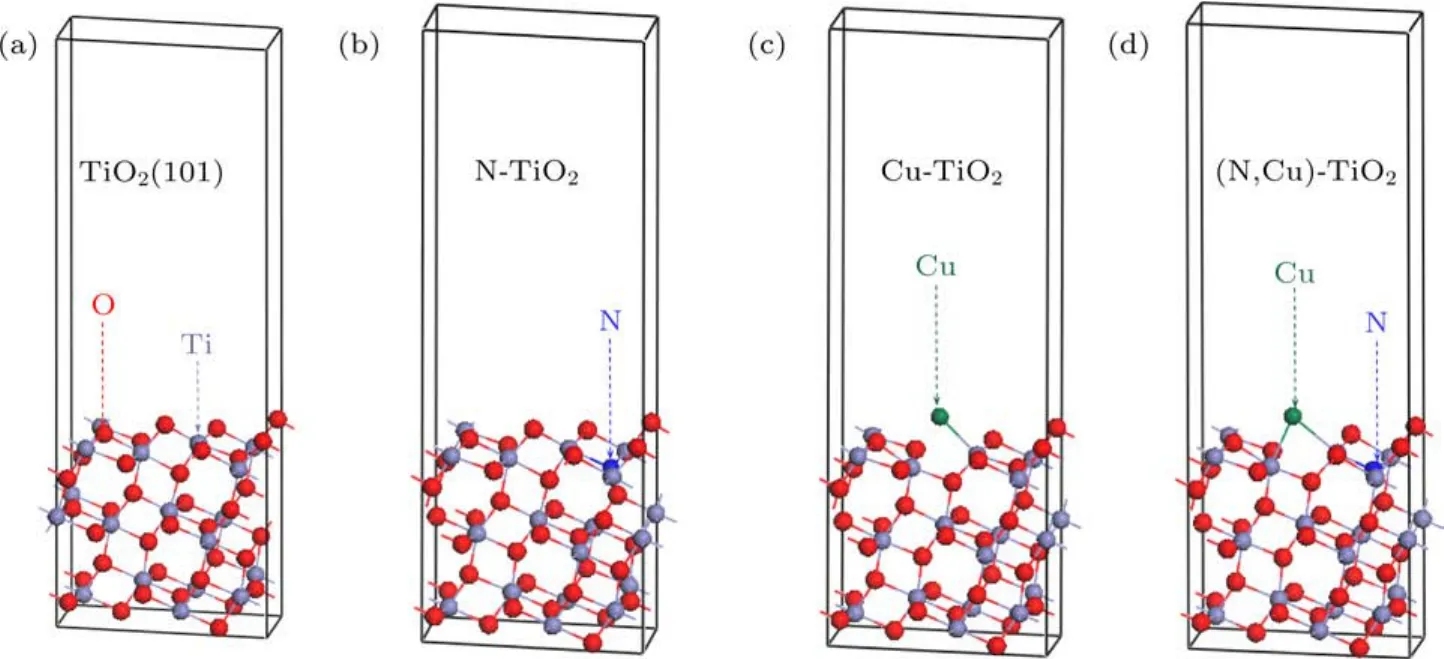
圖4 優化后的TiO2(101)表面的超胞模型 (a)純TiO2;(b)N摻雜TiO2(N@O);(c)Cu摻雜TiO2(Cu@O);(d)(Cu,N)共摻雜TiO2(Cu@O&N@O)Fig.4 .Optimized supercell models of anatase TiO2(101)surface:(a)Pure TiO2;(b)N doped TiO2(N@O);(c)Cu doped TiO2(Cu@O);(d)(Cu,N)codoped TiO2(Cu@O&N@O).
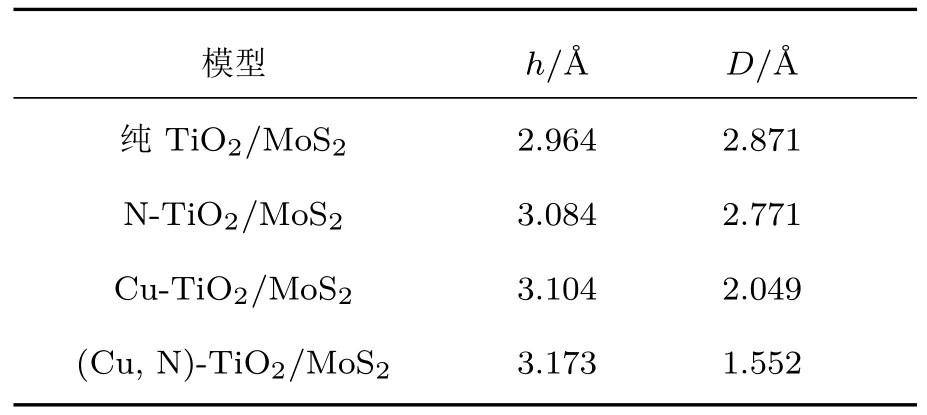
表2 單層MoS2的厚度(h)和單層MoS2與Cu/N(共)摻雜TiO2(101)表面之間的垂直距離(D)Table 2 .Thickness(h)of the MoS2and the vertical separation(D)between MoS2and Cu or/and N doped TiO2(101)surface.
3.2 缺陷形成能
為了解不同生長環境下摻雜體系的穩定性,我們根據以下公式計算了不同替位摻雜體系的形成能:
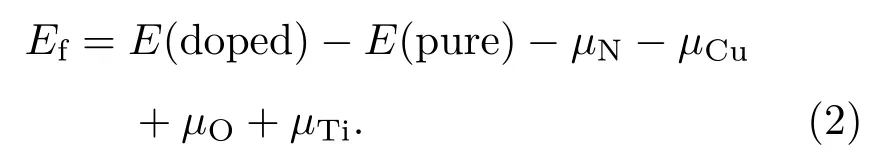
間隙位置摻雜體系的形成能計算公式如下:
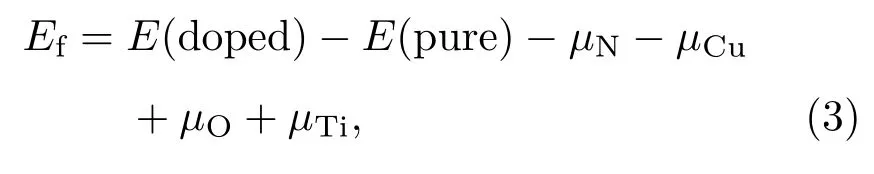
其中E(doped)和E(pure)分別表示摻雜體系和純銳鈦礦TiO2(101)表面的總能量.化學勢μN和μO分別從自由分子N2(μN= μ(N2)/2)和O2(μO= μ(O2)/2)的基態能量中得出,Cu和Ti的化學勢分別從其塊狀材料基態能量得出.然而,形成能計算有富Ti和富O兩種情況[41].結構中Ti和O的化學勢滿足μTi+2μO= μ(TiO2),在富Ti條件下,μTi取塊體Ti的化學勢,O的化學勢為μO=(μ(TiO2)? μTi)/2. 在富O條件下,μO取O2分子的基態能量(μO= μ(O2)/2),μTi= μ(TiO2)? μ(O2).
不同摻雜情況下缺陷形成能計算結果如表3所示,Ef越小就意味著雜質原子越容易在TiO2(101)表面摻雜.在選擇替位摻雜位置時,計算結果顯示替換TiO2(101)表面頂層原子(靠近真空層位置)的形成能相比于替位內部原子(遠離真空層位置)都稍小一些,因此,替位頂層原子的結構更容易形成.而TiO2(101)表面的頂層存在配位數分別為2和3的兩種O原子,通過分別計算其缺陷形成能得出Cu和N替位配位數分別為2和3時的O原子時形成能最小.因此,下文中的Cu@O,N@O體系指所替位的O原子的配位數分別為2和3的頂層氧原子的摻雜體系.對于Cu/N(共)摻雜的TiO2(101)表面,在富Ti生長條件下,Cu@O和N@O的缺陷形成能遠小于其他摻雜體系,其幾何形狀如圖4(b)和圖4(c)所示.在富O的生長環境下,Cu@Ti的缺陷形成能遠小于其他摻雜體系.結果表明,在富Ti條件下,O離子更容易被N和Cu雜質取代,而在富O條件下,Ti離子更容易被Cu雜質取代.對于Cu和N共摻雜的TiO2(101)表面(表3),在富Ti生長條件下,Cu@O和N@O的缺陷形成能遠小于其他共摻雜位置的體系,幾何結構如圖4(d)所示.尤其在富Ti條件下,Cu@O和N@O的缺陷形成能為?9.146 eV,表明Cu@O和N@O的替位摻雜很容易形成.同樣,對于Cu或/和N摻雜的TiO2/MoS2異質結,在富Ti條件下,Cu@O和N@O的缺陷形成能為最小,表明Cu@O和N@O很容易在異質結中形成.因此本文選取Cu和N替代O的結構來計算結構的相關性質.
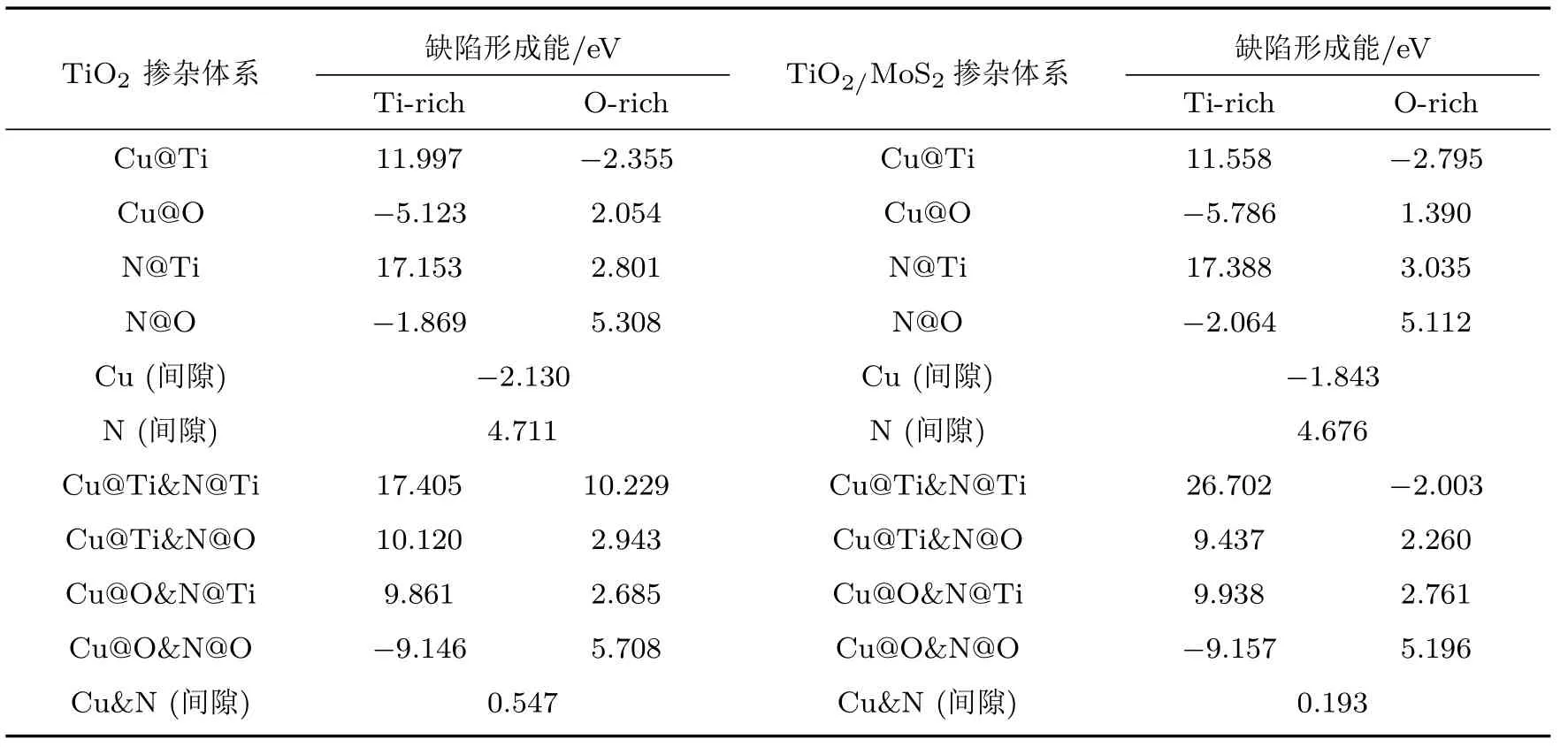
表3 Cu/N(共)摻雜的銳鈦礦TiO2(101)表面和Cu/N(共)摻雜的TiO2/MoS2異質結的缺陷形成能Table 3 .Defect formation energy of Cu or/and N doped anatase TiO2(101)surface and TiO2/MoS2 heterostructure.
3.3 電子態密度和電荷轉移分析
為了研究Cu/N(共)摻雜對TiO2/MoS2異質結光催化活性和光吸收性能的影響,通過雜化泛函計算了Cu/N(共)摻雜的TiO2(101)表面和Cu/N(共)摻雜TiO2/MoS2異質結的態密度,結果如圖5所示.
圖5描述了所有體系的總態密度(total density of states,TDOS)和對應的所有體系分波態密度(partial density of states,PDOS).由圖5(a)和圖5(a′)可以發現,TiO2(101)表面的帶隙為2.90 eV,其價帶頂(value based management,VBM)主要由O 2p態提供,導帶底(conduction band minimum,CBM)主要由Ti 3d態組成,這與實驗結果相符合,表明本文計算結果的正確性.與純的TiO2(101)表面態密度相比,N摻雜后對TiO2(101)表面的導帶和價帶位置影響不大,但在帶隙中存在N 2p的雜質態(圖5(b)和圖5(b′)),這種雜質態可以增強材料在可見光范圍的光吸收,提高材料對可見光的利用.Cu摻雜體系的禁帶中存在Cu 3d雜質態(圖5(c)和圖5(c′)),同樣能夠提高材料的光催化活性.由圖5(d)和圖5(d′)得出在(Cu,N)共摻雜的TiO2(101)表面中,由于Cu和N的協同作用,使得體系的帶隙明顯減小,這樣能有效提高材料對可見光的利用率.

圖5 (共)摻雜體系的TDOS和PDOS圖 (a)和(a′)純的TiO2(101)表面;(b)和(b′)N摻雜的TiO2(101)表面;(c)和(c′)Cu摻雜的TiO2(101)表面;(d) 和(d′)(N,Cu)共摻雜TiO2(101)表面;(e)和(e′)純的TiO2/MoS2;(f)和(f′)N-TiO2/MoS2;(g)和 (g′)Cu-TiO2/MoS2;(h)和 (h′)(N,Cu)-TiO2/MoS2(費米能級設置為 0)Fig.5 .Calculated TDOS and PDOS of(co)doping systems:(a)and(a′)Pure TiO2(101)surface;(b)and(b′)N doped TiO2(101)surface;(c)and(c′)Cu doped TiO2(101)surface;(d)and(d′)(N,Cu)doped TiO2(101)surface;(e)and(e′)pure TiO2/MoS2;(f)and(f′)N-TiO2/MoS2;(g)and(g′)Cu-TiO2/MoS2;(h)and(h′)(N,Cu)-TiO2/MoS2(The Fermi level is set to zero).
關于TiO2/MoS2異質結的態密度,計算結果表明TiO2/MoS2異質結的帶隙(1.38 eV,見圖5(e))比純的TiO2(101)表面帶隙(2.90 eV)明顯減小.這意味著在TiO2/MoS2異質結中電子能容易地從價帶轉移到導帶,導致光吸收能力提高.在Cu和N摻雜TiO2/MoS2異質結后,態密度的結果如圖5(f)和圖5(g)所示,體系的帶隙有所減小,但變化不明顯.當(Cu,N)共摻雜TiO2/MoS2異質結時(圖5(h)),由于Cu和N協同作用,在帶隙中費米面附近出現了Cu 3d軌道提供的雜質態,這種雜質態可以起到橋梁作用,使低能光生電子從價帶先躍遷到雜質帶再到導帶,從而增強對低能光子的吸收,可以促進電子空穴對分離,提高材料對可見光的利用率.
為了更清晰地了解Cu/N(共)摻雜異質結體系中TiO2(101)表面和MoS2單層之間的電荷轉移,計算了Cu/N(共)摻雜TiO2/MoS2異質結的差分電荷密度.計算三維差分電荷密度的公式如下:
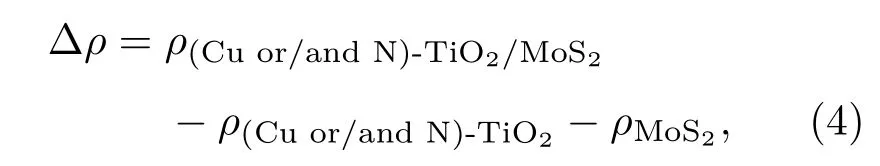
其中ρ(Cu or/and N)-TiO2/MoS2,ρ(Cu or/and N)-TiO2和ρMoS2分別表示Cu/N(共)摻雜的TiO2/MoS2異質結、Cu/N(共)摻雜的TiO2(101)表面和單層MoS2的電荷密度.除此之外,通過沿x-y平面的積分電子密度差法計算了平面平均差分電荷密度,公式如下:

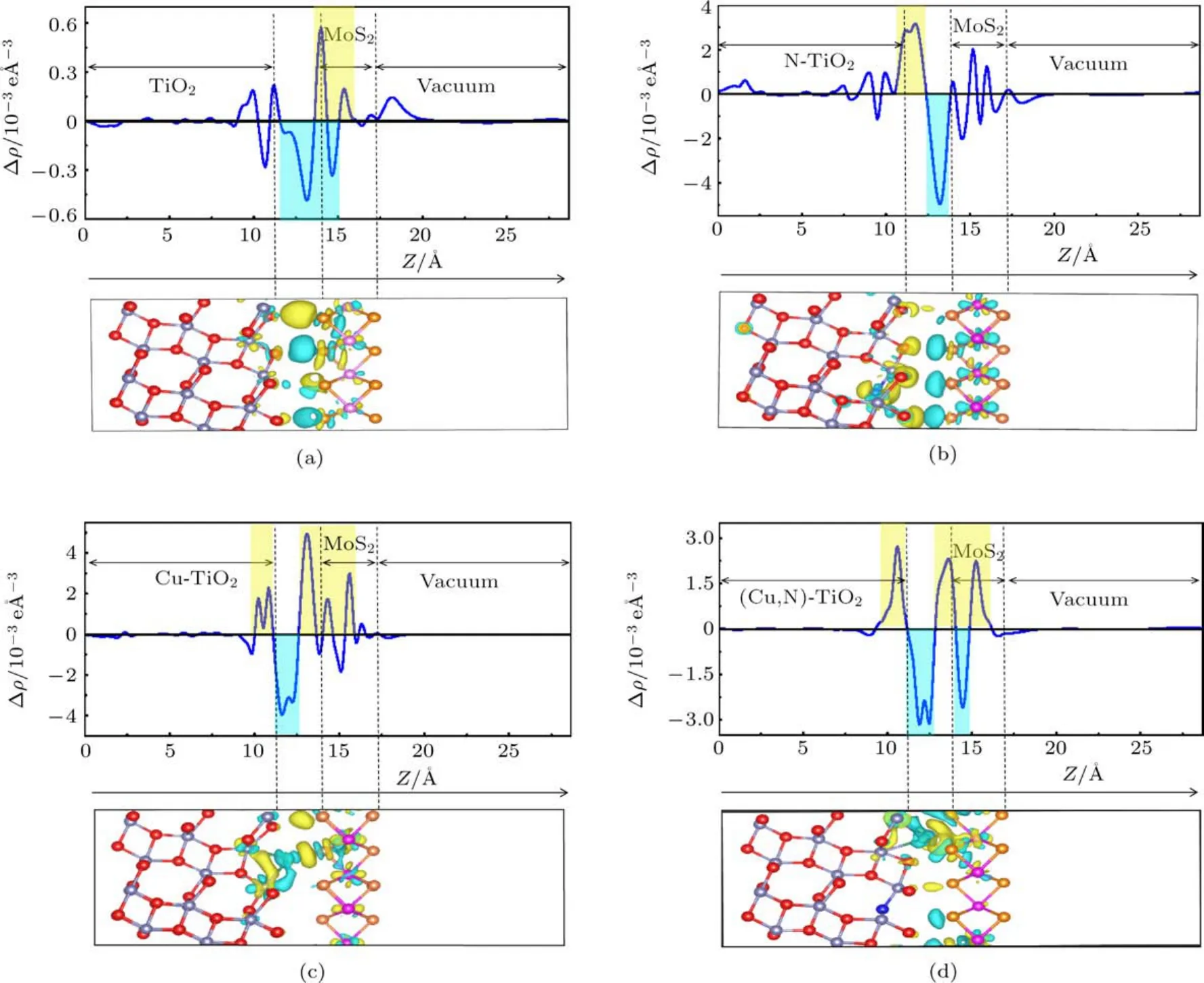
圖6 (a)純TiO2/MoS2,(b)N-TiO2/MoS2,(c)Cu-TiO2/MoS2,(d)(Cu,N)-TiO2/MoS2的差分電荷密度圖(每個分圖上面的是z方向上的平面平均差分電荷密度,正值表示電子積累,負值表示電子耗盡;下面是三維差分電荷密度圖,黃色和青色區域分別表示電子積累和耗盡)Fig.6 .Charge density dif f erence:(a)Pure TiO2/MoS2;(b)N-TiO2/MoS2;(c)Cu-TiO2/MoS2;(d)(Cu,N)-TiO2/MoS2(Top of a function of position in the z-direction,the positive value indicates electron accumulation,and the negative value indicates electron depletion.Bottom of three dimensional charge density dif f erence,the yellow and cyan areas indicate electron accumulation and depletion,respectively).
其中z軸是沿界面法線的方向,x-y平面是垂直于z軸的超截面;i和j分別表示a軸和b軸格.計算結果如圖6所示.
圖6中分別描述了TiO2/MoS2體系、 NTiO2/MoS2體系、Cu-TiO2/MoS2體系和(Cu,N)-TiO2/MoS2體系平面和三維差分電荷密度圖.在平面差分電荷密度圖中,正值方向表示電荷積累,負值方向表示電荷耗盡.在三維差分電荷密度圖中,青色區域表示電荷耗盡,黃色區域表示電荷累積.三維差分電荷密度圖表明,在Cu/N(共)摻雜的TiO2(101)表面和MoS2單層之間電荷發生轉移.圖6(a)TiO2/MoS2體系差分電荷密度圖可以看出,電荷由TiO2(101)表面轉移向MoS2單層.由圖6(b)可以看出,當N原子摻雜TiO2/MoS2異質結后,電荷由MoS2轉移向TiO2(101)表面,并且電荷轉移量也有所增加,這是N原子摻雜引起了電荷轉移的變化.在Cu摻雜和(Cu,N)共摻雜的TiO2/MoS2異質結中,電荷由異質結界面附近的原子分別轉移向TiO2和MoS2兩側(圖6(c)和圖6(d)),這可能是由于Cu原子附近的電荷發生轉移引起的.以上結果表明,本文研究的幾種摻雜能夠有效提高復合體系電荷轉移能力,抑制光生電子和空穴的復合,從而提高材料的光催化活性.
3.4 光學性質
作為光催化劑,高效吸收太陽光是它的基本需求,尤其是對可見光的吸收.為了探索對可見光吸收的機理,通過理論計算了材料對紫外和可見光吸收光譜.為了很好地比較,通過偶極子近似中的費米黃金法則,計算了純TiO2(101)表面、單層MoS2,Cu/N(共)摻雜的TiO2(101)表面和Cu/N(共)摻雜的TiO2/MoS2異質結的頻率相關介質矩陣.介電函數的虛部表達式如下:
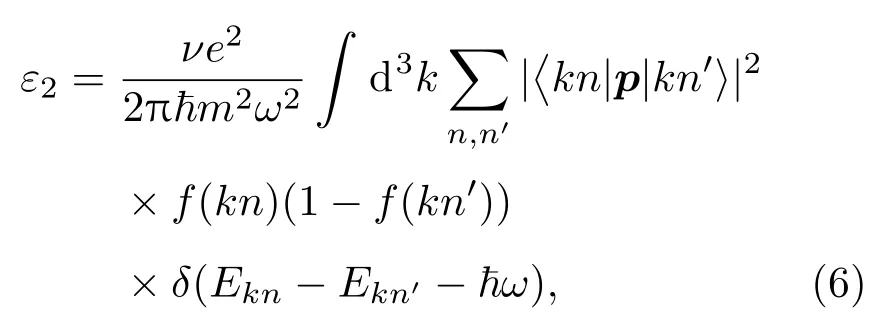
其中 ~ω是入射光子的能量,代表動量算符;r(h/i)(?/?x)是電子波函數;f(kn)是費米函數.通過Kramer-Kronig變換從虛部求出介電函數的實部. 吸收系數I(ω)由ε1(ω)和ε2(ω)導出, 公式如下:

考慮到介電函數的張量性質,ε1和ε2取沿x,y,z方向的平均值.而在計算吸收系數時,按照目前VASP程序的理論,考慮了帶間躍遷,例如在(6)式中,計算虛部時涉及到帶間躍遷的kn和kn′兩個態.上述關系是能帶結構和光學性質分析的理論基礎,反映了不同能級之間電子躍遷引起的吸收光譜機理.
半導體光催化劑的光學性質是其光催化性能的關鍵因素之一. 因此,本文詳細計算了TiO2(101)表面和Cu/N(共)摻雜的TiO2(101)表面的吸收光譜,結果如圖7所示.圖7(a)描述了Cu/N(共)摻雜的TiO2(101)表面的光吸收圖,并將計算結果與實驗吸收光譜[42](圖7(b))進行了比較.圖7(c)中包括了TiO2/MoS2異質結、Cu/N(共)摻雜的TiO2/MoS2異質結的吸收光譜.單層MoS2,TiO2(101)表面和TiO2/MoS2異質結的吸收光譜如圖7(d)所示.
結果表明,由于銳鈦礦型TiO2(101)表面的禁帶寬度(2.90 eV)較大,所以它只能吸收紫外光,而對可見光不能有效利用.通過給TiO2(101)表面摻雜能夠有效改善這一缺陷.
例如N摻雜TiO2(101)表面后,體系的帶隙變為2.58 eV,在近紫外光區域中和可見光(λ<500 nm)區域光吸收效率明顯增加.對于Cu摻雜的TiO2(101)表面,Cu 3d軌道影響體系的禁帶寬度,使體系在可見光區域內具有良好的光吸收率.對于(Cu,N)共摻雜TiO2(101)表面,Cu和N雜質共摻雜的協同效應導致禁帶寬度縮小,使體系對可見光的吸收大大增強.結果可能是(Cu,N)共摻雜TiO2(101)表面導致可見光光催化活性提高和光吸收邊緣明顯紅移.計算結果與實驗數據相符合(圖7(b)).為了提高TiO2(101)表面的光催化活性,構建出TiO2/MoS2異質結.計算TiO2/MoS2異質結的光吸收性能,結果如圖7(d)所示.從圖7(d)可以看出,TiO2/MoS2異質結相比TiO2(101)表面的可見光來說,其光催化活性提高了,光吸收邊緣明顯發生了紅移.結果表明,構建異質結能夠有效提高材料對太陽光的利用.為了進一步提高材料對太陽光的利用,我們構造Cu/N(共)摻雜到TiO2/MoS2異質結中TiO2(101)表面的結構,計算出各種結構的光吸收性能,結果如圖7(b)所示.與TiO2/MoS2異質結的光學性質相比,由于N 2p軌道影響,N-TiO2/MoS2體系在近紫外光區域和可見光(λ<630 nm)區域中的光吸收性能明顯增加.計算Cu-TiO2/MoS2體系的光催化性能,結果表明它在可見光范圍內的光吸收略有增強.相比于單摻雜體系,(Cu,N)共摻雜TiO2/MoS2體系對于太陽光的吸收能力有著顯著的提高.總而言之,異質結的形成和Cu/N(共)摻雜到TiO2/MoS2異質結都可以增強光吸收,并改變吸收邊緣.
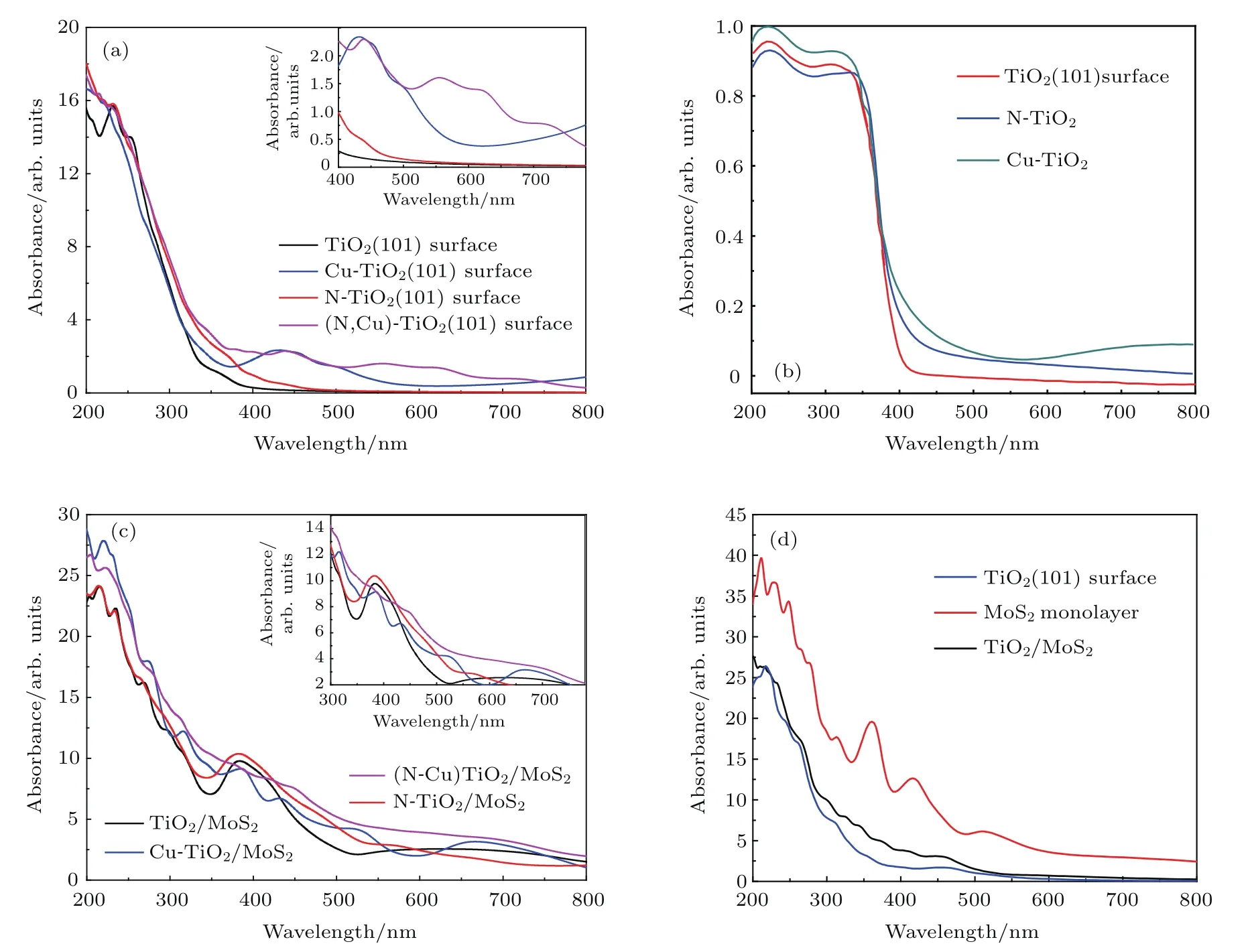
圖7 (a)摻雜TiO2(101)表面的光學吸收光譜;(b)摻雜TiO2(101)表面的光學吸收光譜的實驗數據;(c)摻雜TiO2/MoS2異質結的光學吸收光譜;(d)TiO2(101)表面、單層MoS2和TiO2/MoS2異質結的光學吸收光譜Fig.7 .(a)Optical absorption spectra of doping TiO2(101)surface;(b)experimental data of optical absorption spectra of doping TiO2(101)surface;(c)optical absorption spectra of doping TiO2/MoS2heterostructure systems;(d)optical absorption spectra of TiO2(101)surface,MoS2monolayer and TiO2/MoS2heterostructure systems.
3.5 壓力對TiO2/MoS2異質結的影響
在以上研究的基礎上,繼續研究了壓強對TiO2/MoS2異質結復合材料光催化性能的影響.采用的模型為優化好的TiO2/MoS2異質結.對TiO2/MoS2異質結分別施加10,20,30,40,50 GPa的靜水壓,并計算在不同壓力下的電子結構和光學性質.結果發現,壓強增加導致體系的晶格常數以及體系的真空層都逐漸減小,整個結構均處于壓縮狀態.
圖8(a)中給出了體系總能量隨壓強變化的關系,從圖8(a)中可以看出,從10 GPa到50 GPa體系總能隨壓強線性增長,這說明體系隨著壓強的增加穩定性在減弱.因為在TiO2/MoS2異質結幾何結構優化后,晶體結構中的各原子都達到了最穩定的平衡狀態,總能量達到最低狀態,但隨著外界壓強的增大,晶體勢能逐漸增大,從而體系的總能量也增加.圖8(b)和圖8(c)分別描述了體系的態密度和光學性質.從圖8(b)中可以發現,隨著壓強的增加體系禁帶寬度逐漸減小,但不同范圍的壓強對帶隙影響大小不一樣.例如,在體系分別加0 GPa和10 GPa的壓強時,兩狀態體系的禁帶寬度無明顯變化,說明體系加10 GPa的壓強對體系電子性能影響不大.當體系施加的壓強逐漸從10 GPa增加到30 GPa時,體系帶隙明顯減小(0.15 eV).然而當給體系施加壓強增加到40 GPa時,體系禁帶寬度相對于30 GPa出現明顯減小(0.35 eV),這可能是由于體系發生相變導致.當壓強增加到50 GPa時,體系的帶隙變為0.30 eV.這表明,通過加壓可以有效地減小體系的帶隙.從圖8(c)中可以看出,當給體系施加10 GPa的壓強后,復合材料對近紫外區的吸收能力有顯著的提高,并對400—700 nm范圍的可見光吸收能力也有提高,吸收波峰向可見光轉移.隨著對復合材料繼續加壓,復合體系對近紫外和可見光的吸收性能依次提高.當對復合體系的壓強施加到50 GPa時,復合體系對450—800 nm區域的光吸收能力有明顯提高.這表明,通過給復合材料加壓可以有效提高材料的光學性質.

圖8 TiO2/MoS2異質結在不同壓強下的(a)結合能,(b)光吸收譜和(c)態密度Fig.8 .(a)Formation energy diagram,(b)optical absorption map and(c)density of state of TiO2/MoS2 heterostructure at dif f erent pressures.
4 結 論
本文采用雜化密度泛函方法對TiO2/MoS2異質結、Cu/N(共)摻雜的TiO2(101)表面和Cu/N(共)摻雜的TiO2/MoS2異質結結構的電子結構和光學性質進行了研究.結果表明,TiO2/MoS2異質結比純TiO2(101)表面的帶隙明顯減小,(Cu,N)共摻雜誘導TiO2/MoS2異質結帶隙中出現了N 2p和Cu 3d雜質態,減小了光生電子-空穴的復合率,提高了材料對可見光的吸收能力.計算了Cu/N(共)摻雜的TiO2/MoS2異質結的差分電荷密度,發現電荷最終會由異質結的一側轉移到另一側,有效抑制了光生電子-空穴的復合.因此,Cu/N(共)摻雜的TiO2/MoS2異質結光催化劑具有令人滿意的光催化活性.除此之外,計算了壓力對復合材料光催化性能的影響,發現通過施加壓力可以有效提高材料的光催化活性.這些理論計算可以為實驗提供一個引導與理論解釋.
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
