高效液相色譜-質譜聯用法測定肥料中赤霉酸含量的不確定度評估*
2018-10-26 10:07:16段路路王高俊
肥料與健康 2018年4期
關鍵詞:標準
段路路,王高俊,嚴 虎
(上海化工研究院有限公司/上海化學品公共安全工程技術研究中心 上海 200062)
植物生長調節劑是由人工合成的化學物質或從生物中提取的天然植物激素,具有調節植物生長和發育的功能[1]。目前,世界上公認的植物生長調節劑有五大類,即生長素類、赤霉素類、細胞分裂素類、乙烯和脫落酸類[2]。其中,赤霉酸是一種廣譜性植物生長調節劑,具有促進植物細胞分裂和種子發育、誘導花芽生長、提高瓜果植物的坐果率或促進無核果實的形成、葉菜類植物的營養生長等作用,也具有水果保鮮劑的作用。由于植物生長調節劑在較低的濃度下即可對植物的生長發育表現出良好的調節作用,因此,在農業生產中合理使用植物生長調節劑具有很好的效果[3]。到目前為止,我國批準登記的植物生長調節劑品種有38種共587個產品[4]。隨著我國農業生產技術的快速發展,運用植物生長調節劑調控植物的生長和產量,已逐漸成為農業生產中必不可少的技術手段[5]。
肥料是農業生產中極為重要的生產資料,是保障國家糧食安全的重要物質基礎,其質量直接影響農業生產及糧食安全。植物生長調節劑作為一種常用的農藥產品,目前在全世界農業生產中得到了廣泛應用,但不允許在肥料中隨意添加。然而在高額利潤的誘惑下,個別肥料生產商違規添加促進作物生長的調節劑,以增強肥料的肥效。近年來,一些不法肥料生產者及經營者,為了追求立竿見影的“肥效”,在肥料中違禁添加植物生長調節劑,對農產品的質量安全造成威脅。農產品中殘留的植物生長調節劑通過食物鏈進入人體后,會對人體造成一定程度的傷害,輕則引起腹瀉等疾病,重則導致人體免疫功能下降、骨骼疏松,甚至出現致畸、致癌、致基因突變等嚴重后果[3]。目前,肥料中隱性添加的赤霉酸既無限量指標要求,又缺乏規范統一的檢測方法標準,嚴重影響政府監管工作的有效開展。隨著肥料的廣泛應用,政府監管部門要做好產品質量監管以及質量安全風險監測工作,國內急需建立肥料中赤霉酸的檢測標準和方法,以規范行業行為,促進肥料產業健康發展。因此,建立肥料中赤霉酸含量的檢測方法意義重大。
目前,肥料中赤霉酸的常用檢測方法有高效液相色譜法[6]、高效液相色譜-質譜聯用法(以下簡稱液質聯用法)[1]等,但無論采用何種方法檢測,測定結果都有一定的不確定度[7]。不確定度評定是檢測結果的重要部分,反映了檢測結果的可靠性以及檢測過程中各項不確定度來源對測定結果的影響程度。為了表征檢測工作和結果的準確性,實驗室通常采用不確定度來對檢測結果進行說明[8- 9]。
由于液質聯用法具有靈敏度高、定性定量準確等特點,同時目前針對肥料中赤霉酸含量檢測的不確定度評定研究較少,在參考《測量不確定度評定與表示指南》[10]中的理論依據和建立不確定度數學模型的步驟以及其他相關文獻資料[11]的基礎上,采用液質聯用法對肥料中的赤霉酸含量進行測定,對實驗室采用的檢驗方法、所使用的儀器設備、標準物質及其溶液、標準曲線、樣品處理過程等影響試驗結果的因素進行不確定度評定[12- 13],以期找到影響測定結果可靠性的主要因素,從而對試驗過程進行有效監控,保證最終數據的準確性和可靠性。
1 材料與方法
1.1 材料與試劑
赤霉酸標準品,質量分數98.0%,德國Dr. Ehrensforfer公司;甲醇,色譜純,德國Merk公司;甲酸,分析純,中國國藥集團化學試劑有限公司;超純水,采用Mili- Pore超純水系統制備;肥料樣品,市售,水溶性肥料;初始流動相,甲酸-甲醇溶液(準確移取1.0 mL甲酸,用甲醇稀釋至1 L)和甲酸溶液(準確移取1.0 mL甲酸,用水稀釋至1 L)按體積比1∶9混合均勻,現配現用。
1.2 儀器與設備
4500QTRAP三重四級桿質譜聯用儀,美國AB公司;Agillent 1290高效液相色譜儀,美國Agillent公司;Mili- Pore超純水系統,密理博公司;AL204- 1C電子天平,瑞士梅特勒-托利多公司;1 000 μL移液器和10 mL移液器,艾本德中國有限公司;漩渦混合器,上海安譜實驗科技股份有限公司;超聲波清洗器,上海科導超聲儀器公司;高速離心機,上海盧湘儀離心機儀器有限公司。
1.3 試驗方法
1.3.1 標準儲備溶液和使用溶液的配制
準確稱取赤霉酸標準品0.1 g(精確至0.1 mg)于100 mL容量瓶中,用甲醇溶解并稀釋至刻度,得到的赤霉酸標準儲備溶液質量濃度為1 mg/mL。
用移液器吸取100 μL赤霉酸標準儲備溶液于100 mL容量瓶中,用甲醇稀釋至刻度,得到的赤霉酸標準溶液質量濃度為1 μg/mL。
用移液器準確移取0、100、200、500、1 000和2 000 μL赤霉酸標準溶液分別置于10 mL容量瓶中,用初始流動相定容,得到質量濃度分別為0、10、20、50、100和200 μg/L的赤霉酸標準工作溶液。
1.3.2 樣品前處理
稱取1~2 g(精確至0.1 mg)樣品于50 mL離心管中,加入20 mL流動相,用旋渦混合器均質1 min后于室溫下超聲30 min,然后置于轉速為8 000 r/min的離心機內離心5 min,轉移全部上層清液至25 mL容量瓶并用初始流動相定容,搖勻后取適量樣品溶液經0.22 μm有機相微孔濾膜過濾,待測。
1.3.3 分析條件
試驗選用InfinityLab Poroshell 120 SB- C18色譜柱(3.0 mm×50.0 mm,2.7- Micron);流動相A為甲酸-甲醇溶液,流動相B為甲酸溶液;進樣量為5 μL。
質譜采用電噴霧電離源(ESI),多反應監測(MRM)模式,定量離子為345.1/143.1,定性離子為345.1/239.2,離子化電壓為-4 500 V,離子源溫度為500 ℃,去簇電壓為-50 V,碰撞電壓為-20 V。流動相梯度洗脫程序如表1所示。
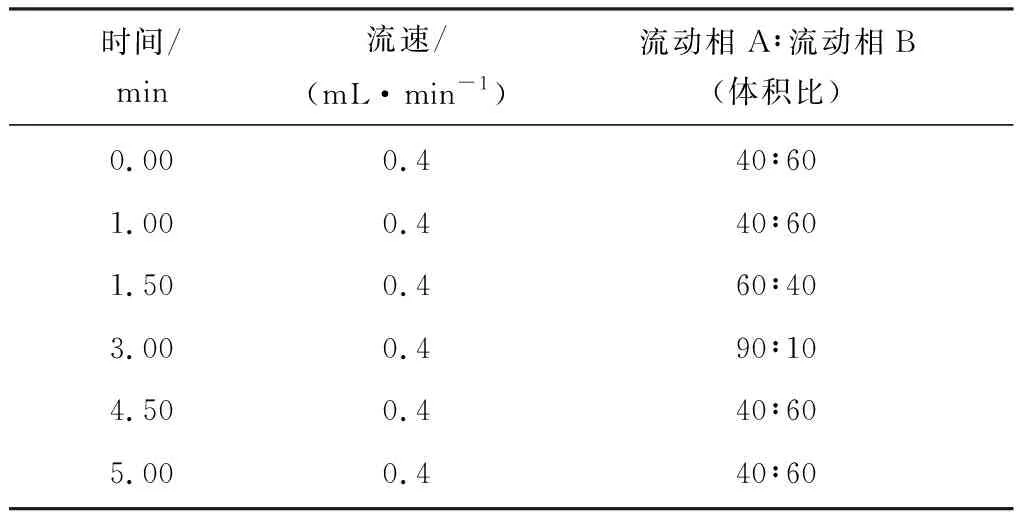
表1 流動相梯度洗脫程序
1.3.4 數學模型的建立
試樣中赤霉酸的含量按式(1)計算:
(1)
式中:W——試樣中赤霉酸的含量,mg/kg;
C——試樣溶液中赤霉酸的實測質量濃度,mg/L;
V——樣品定容體積,mL;
f——樣品溶液的稀釋因子;
m——樣品稱樣量,g。
根據檢測過程及數學模型,可判斷影響測定結果不確定度的來源有樣品重復測定、標準曲線擬合、樣品及標準物質稱量、標準物質配制、樣品定容以及標準物質純度等,如圖1所示。
2 結果與分析
2.1 標準溶液及配制產生的相對不確定度urel(C)
2.1.1 標準儲備溶液引入的相對不確定度urel(儲)


圖1 不確定度來源分布


則標準儲備溶液引入的相對不確定度:

=0.006 86。
2.1.2 標準溶液配制過程產生的不確定度
2.1.2.1 容量瓶引入的相對不確定度urel(V容)
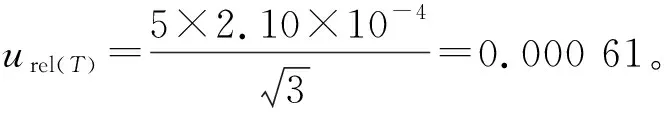
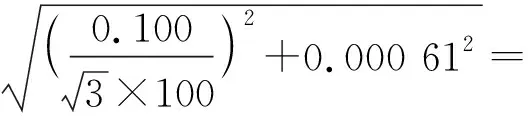

2.1.2.2 移液器引入的相對不確定度urel(V器)

式中:ti——容量允許誤差;
V器——移液器規格,μL。
各可調移液器的容量允許誤差及標準不確定度計算結果如表2所示。

表2 各可調移液器的容量允許誤差及標準不確定度計算結果
由移液器引入的合成相對不確定度urel(V器)=(0.001 152+0.000 582+0.000 582+0.000 582+0.000 062)0.5=0.001 53。
2.1.3 標準溶液及配制產生的相對不確定度urel(C)
根據上述研究,赤霉酸標準溶液配制過程中產生的相對不確定度包括赤霉酸標準儲備溶液配制過程引入的相對不確定度、所使用的容量瓶及移液器引入的相對不確定度,上述標準溶液配制產生的相對不確定度:

=0.007 11。
2.2 樣品測定產生的相對不確定度urel(S)
2.2.1 樣品稱量產生的相對不確定度urel(m樣)

2.2.2 樣品前處理產生的相對不確定度urel(S前)

2.2.3 樣品重復性測定引入的相對不確定度urel(S重)
根據液質聯用的分析條件,對肥料樣品進行了10次測定,測定結果如表3所示。
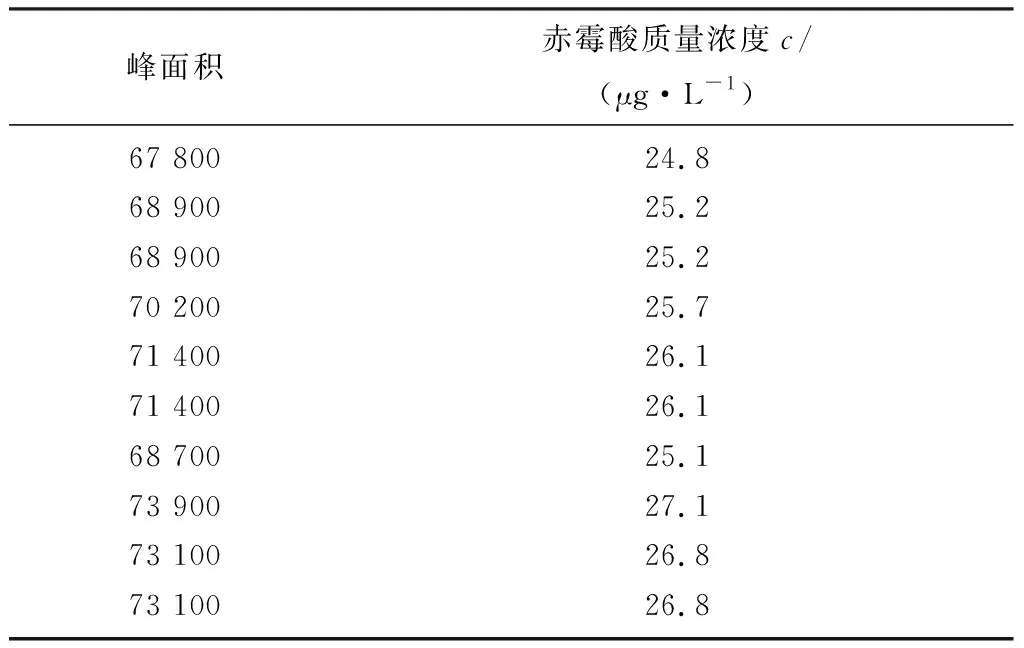
表3 肥料樣品中赤霉酸濃度測定結果
因此,樣品重復性測定的標準不確定度uS重=S(c)=0.817g/L,樣品重復性測定的相對不確定度
2.2.4 樣品測定產生的相對不確定度urel(S)
根據上述研究,樣品中赤霉酸測定產生的不確定度包括樣品稱量產生的不確定度、樣品前處理產生的不確定度以及樣品重復性測定引入的不確定度,上述樣品測定產生的相對不確定度:

=0.031 55
2.3 線性擬合過程引入的測量相對不確定度urel(c線)


表4 標準工作溶液測量結果
根據表4數據,可計算得出峰面積測量的標準偏差為:
式中:yi——由儀器檢測各點的相應峰面積;
yfi——由標準曲線方程計算得到的峰面積;
n——配制5個標準工作溶液,每個濃度點測定3次,共計15次,即n=15。
當對樣品進行測量時,被測量的樣品濃度是由峰面積通過最小二乘法擬合的線性方程得到,因此由標準曲線擬合引入的測量結果的標準不確定度為:

式中:b——擬合直線的斜率;
p——試樣平行測定次數,p=10;


xi——標準工作溶液的質量濃度,μg/L。
2.4 液質聯用儀校準引起的相對不確定度urel(Q)

2.5 相對不確定度urel(X)的合成
液質聯用法測定肥料樣品中赤霉酸含量的相對不確定度主要包括標準溶液及配制產生的相對不確定度urel(C)、樣品測定產生的相對不確定度urel(S)、線性擬合過程引入的測量相對不確定度urel(c線)和液質聯用儀校準引起的相對不確定度urel(Q),將上述各分量合成相對不確定度urel(X)為:
=(0.007 112+0.031 552+0.050 182+
0.011 552)0.5=0.060 81
2.6 擴展不確定度及測定結果報告
95%置信概率下取包含因子k=2,將合成相對不確定度乘以包含因子計算得到的肥料樣品中赤霉酸含量測定結果的擴展不確定度uX=urel(X)×k=26.3×2=52.6(μg/kg), 故液質聯用法測定肥料中赤霉酸含量的結果報告表示為(432±52.6)μg/kg。
3 結果與討論
通過建立液質聯用法測定肥料中赤霉酸含量的不確定度評價模型,從而保證肥料中赤霉酸含量測定結果的有效性和合理性,為液質聯用法測定肥料中赤霉酸含量的質量控制提供了有效、可靠的測量體系。
結果表明,肥料中赤霉酸的含量為(432±52.6)μg/kg,k=2。從各相對不確定度分量的貢獻率分析(表5)可知,試驗過程中,測量不確定度主要來源于兩個方面,即樣品濃度測定中標準曲線的擬合引入的相對不確定度和樣品重復測定引入的相對不確定度,而標準物質純度、標準物質和樣品的稱量、樣品前處理的相對不確定度影響不大。因此,在測定過程中,檢測人員應掌握實驗操作技能,增加標準曲線的濃度水平數以減少由擬合曲線帶來的影響,并增加平行測量次數以減少測試重復性對結果的影響;同時,加強設備的日常維護和管理,盡可能保證試驗結果的穩定性和準確性。

表5 各相對不確定度分量的貢獻率數據
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
