奧拉西坦的合成工藝改進
2018-08-22 03:10:06吳福鴻李兆林楊亞軍
石油化工應用 2018年7期
吳福鴻,李兆林,楊亞軍,李 飛
(寧夏康亞藥業股份有限公司,寧夏銀川 750002)
奧拉西坦(oxiracetam,1)化學名為 2-(4-羥基吡咯烷-2-酮-1-基)-乙酰胺,結構式(見圖1)。
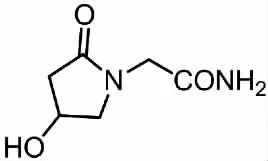
圖1 奧拉西坦的結構式Fig.1 Structural of oxiracetam
奧拉西坦屬吡拉西坦類似物,是一種合成的羥基氨基丁酸(GABOB)環狀衍生物,首次由意大利史克比切姆公司于1974年合成,并于1987年在意大利上市。是新一代腦代謝改善藥,臨床上用于治療健忘癥、老年性癡呆、血管性癡呆等,其藥效活性是吡拉西坦的3~5倍,且耐受性好,不良反應較少[1,2]。
奧拉西坦的合成方法已有不少文獻報道,主要有以下4種:
(1)以亞胺二乙酸乙酯為起始原料,與2-乙氧羰基乙酰氯酰化后再經環合、水解、還原得2-(4-羥基-2-氧代吡咯烷-1-基)乙酸乙酯,最后氨解得1[3]。該法路線較長,合成2-(4-羥基-2-氧代吡咯烷-1-基)乙酸乙酯時成分較復雜且須經柱色譜分離純化,產品總收率低,成本較高且操作繁瑣,不利于工業化生產。
(2)以雙乙烯酮為起始原料,經氯化、酯化得(E)-4-氯-3-甲氧基-2-丁烯酸甲酯,再與甘氨酸環合、酯化得4-甲氧基-吡咯啉-2-酮-1-乙酸甲酯,最后經氨化、還原制得1,總收率22.5%[4]。該法涉及羥基的保護和脫保護步驟,無形中增加成本,操作繁瑣,且收率較低。
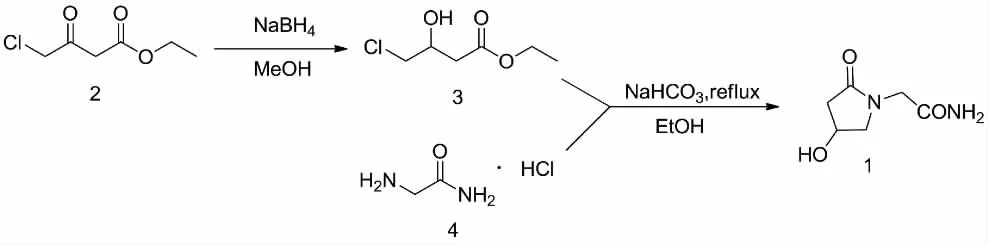
圖2 奧拉西坦的合成路線Fig.2 Synthetic route of oxiracetam
(3)以4-氯-3-羥基丁酸酯為原料,與甘氨酰胺鹽酸鹽直接環合制得1,總收率70.1%[5]。該法雖步驟簡短,但反應時間長達25 h,終產物經硅膠和活性炭脫色后,還需采用離子交換樹脂和重結晶進行純化,起始原料成本高,操作繁瑣。
(4)以4-氯乙酰乙酸酯為原料,經還原、環合反應,制得 1[6]。
以上合成方法存在的主要缺陷有的合成路線長,操作復雜,原料較貴不易得。有的合成路線的原料毒性較大、安全性較低,最終造成產品收率低、成本高、設備投資大、環境污染嚴重等現象,不易適合工業化大生產。尋求一種適合工業化生產的合成方法,對該化合物的深入研究具有重要的意義。作者在深入研究文獻工藝路線的基礎上,以4-氯乙酰乙酸酯為原料,經還原、環合反應,制得1,并通過中試放大工藝研究在原有文獻基礎上有改進和提高。合成路線(見圖2)。
1 實驗
1.1 4-氯-3-羥基丁酸乙酯(3)的合成
于反應瓶中依次加入無水甲醇340.00 g、4-氯乙酰乙酸乙酯(110.40 g,0.66 mol),控制反應液溫度保持在-5 ℃~0 ℃,緩慢加入硼氫化鈉(20.30 g,0.54 mol),調節加料速度,以控制反應體系溫度低于0℃為宜,待硼氫化鈉加畢,保持反應體系溫度為-5℃~0℃,繼續攪拌反應2 h~3 h,TLC跟蹤(展開劑:正己烷-乙酸乙酯=3:1)反應完全;4-氯乙酰乙酸乙酯點消失,判斷反應完全。向反應液中滴加6 mol/L的鹽酸溶液調節反應液pH值為6.0~7.0,攪拌30 min后,減壓濃縮反應液,得棕黃色膠狀濃縮物;濃縮物冷卻至室溫后,向其中加入乙酸乙酯,攪拌30 min后過濾,濾餅用乙酸乙酯淋洗,合并濾液;濾液用純化水洗滌,靜置分層,收集有機層;水層加入乙酸乙酯萃取,收集有機層,合并所得有機層;減壓濃縮至無液體餾出,得淺棕色油狀物;將上述濃縮所得油狀物,于110±10℃進行減壓蒸餾,收集110℃及以上的餾分,得無色透明液體103.40 g,收率:92.6%,GC98.5%(文獻[7]收率 88.05%)。
1.2 2-(4-羥基吡咯烷-2-酮-1-基)-乙酰胺(1)的合成
于反應瓶中依次加入無水乙醇473.00 g,在攪拌狀態下加入甘氨酰胺鹽酸鹽52.50 g(0.48 mol)及碳酸氫鈉 43.90 g(0.52 mol),加畢,升溫 80 ℃~85 ℃至回流;開始向反應液中滴加4-氯-3-羥基丁酸乙酯87.05 g(0.52 mol),分批加入,每滴加約31.60 g 4-氯-3-羥基丁酸乙酯后加入14.60 g碳酸氫鈉,直至4-氯-3-羥基丁酸乙酯和碳酸氫鈉加畢,繼續回流攪拌反應16 h~18 h,采用HPLC法取樣檢測,反應完全,趁熱過濾,濾餅用熱無水乙醇(50℃~60℃)淋洗,棄除濾餅,合并濾液,減壓濃縮,得黃色膠狀濃縮物。向上述濃縮物中加入無水乙醇,于5℃~10℃攪拌析晶2 h~3 h。過濾,濾餅用適量無水乙醇淋洗,濾餅干燥;濾液繼續減壓濃縮后加入無水乙醇,重復上述析晶、過濾處理,棄去濾液,濾餅干燥,合并兩次析晶所得干燥固體,加入到80.00 g純化水加熱至30℃~40℃中攪拌溶解,待固體完全溶解后加入強酸性苯乙烯系陽離子交換樹脂攪拌使溶液pH值為3.0~4.0后,過濾,濾餅用10.00 g純化水洗滌后,回收強酸性陽離子交換樹脂;向濾液中加入強堿性苯乙烯系陰離子交換樹脂攪拌使溶液pH值為7.0,過濾,濾餅用10.00 g純化水洗滌,回收強堿性陰離子交換樹脂;將所得濾液于50℃~60℃、-0.08 MPa~-0.1 MPa條件下減壓濃縮至有大量固體析出時,停止濃縮。向濃縮物中加入300.00 g無水乙醇,攪拌、加熱回流,至固體完全溶解;溶解后加入活性炭5.0 g,攪拌脫色0.5 h后,趁熱過濾,濾餅用20 g~30 g熱乙醇淋洗一次,棄去濾餅,收集濾液。濾液于5℃~10℃下攪拌析晶 2 h~3 h,過濾,濾餅 60±1 ℃干燥 7 h~8 h,得白色固體粗品44.9 g,收率59.1%。粗品經無水甲醇精制得白色結晶性固體40.6 g,收率90.4%,mp 167.8℃~168.5℃。采用高效液相色譜法檢查純度99.96%。ESIMSm/z:181.0[M+Na]+,197.0[M+K]+。1H-NMR(DMSO-d6,400 MHz)δ2.07 (1H,dd,J=2.4&16.8 Hz),2.56(1H,dd,J=6.6&16.8 Hz),3.15 (1H,dd,J=16.2 Hz),4.28(1H,s),5.21(1H,s),7.11(1H,s),7.30(1H,s)。
2 結論與討論
(1)本文以4-氯乙酰乙酸乙酯為起始原料,經還原、環合反應對工藝進行改進,合成的奧拉西坦總收率可達49.5%,較文獻[6]收率有所提高。
(2)對于第一步反應4-氯-3-羥基丁酸乙酯的合成,通過蒸餾收集110℃及以上餾分,將產品提純;再進行第二步反應2-(4-羥基吡咯烷-2-酮-1-基)-乙酰胺的合成,反應原料純度高,且通過分批加入4-氯-3-羥基丁酸乙酯和碳酸氫鈉,使反應在16 h~18 h反應充分。后處理得到的粗品經過無水甲醇一次精制可得高純度的奧拉西坦,對奧拉西坦的質量控制具有重要參考價值。
