GO/Fe3O4/有機胺復合材料的制備及對結晶紫染料的吸附性能
2018-08-01 01:55:02亢璽陽楊清香王立杰宋海媚董夢果陳志軍
無機化學學報 2018年8期
關鍵詞:復合材料
亢璽陽 楊清香 王立杰 宋海媚 張 琰 董夢果 陳志軍
(鄭州輕工業學院,鄭州 450000)
0 引 言
有機染料被廣泛用于多個行業的材料著色,如:紡織、染色、造紙和紙漿、制革和油漆等,這些行業的工業廢水如果不進行治理會含有過量的染料,而有機染料一般難以降解,并對生物體系造成嚴重危害而成為水體污染物[1]。制備能夠吸附有機染料的吸附劑是治理水體中有機染料污染的重要途徑之一,目前吸附劑已成為治理水體污染的研究熱點[2-3]。有多種吸附劑被用于水體中染料分子的吸附移除過程,如:活性炭,殼聚糖,改性硅膠、硅藻土、聚合氧化鋁、灰燼和天然粘土等[4-6]。但是,這些吸附劑存在吸附量小,吸附染料后的吸附劑需要經過長時間靜置沉降才能與水體分離,容易造成二次污染等缺點。因此,研究并開發一種吸附量高并能方便與水體分離的吸附劑,將對水體中染料污染治理具有重要的理論價值和社會經濟價值。石墨烯或氧化石墨烯(GO)已被證實可以在某種程度上移除水體中的有機污染物[7],但存在吸附量較小且難以將其與水體分離而導致嚴重二次污染等缺點[8]。
本文制備了乙二胺 (EDA)功能化的GO/Fe3O4磁性復合材料,乙二胺與丁二胺/己二胺混溶功能化的GO/Fe3O4磁性復合材料,通過胺類有機物對石墨烯的共價鍵修飾,得到結構更加穩定、比表面積更大且具有超順磁性的吸附材料。該材料對結晶紫具有良好的吸附性能,且成本低,可簡單和快速提取/再生,還可以通過外加磁場實現對吸附劑的快速磁分離從而移除水體中的有機染料[9]。
1 實驗部分
1.1 試劑與儀器
濃硫酸購自洛陽市化學試劑廠;石墨粉、高錳酸鉀、氫氧化鈉、雙氧水、三氯化鐵、乙酸鈉、氯化亞鐵、乙二醇、無水乙醇、N,N-二甲基甲酰胺,以上藥品試劑均為天津市風船化學試劑有限公司生產;氯仿、硝酸和鹽酸購自煙臺市雙雙化工有限公司;乙二胺購自天津市德恩化學試劑有限公司;丁二胺、鋰購自阿拉丁試劑有限公司;聚乙烯吡咯烷酮、己二胺購自天津市科密歐化學試劑有限公司;結晶紫購自國藥集團化學試劑有限公司。以上試劑均為分析純,除非特別說明均直接使用。實驗用水為超純水。
樣品的TEM測試儀器為河南省表界面重點實驗室的高分辨透射電子顯微鏡 (型號:JEM-2100UHR),加速電壓為200 kV。樣品的SEM測試是用場發射掃描電子顯微鏡(型號:JSM-7001F),加速電壓為100 kV。使用德國BRUKER公司TENSOR FITR紅外光譜儀,漫反射(DRS)測量,樣品的結構及骨架振動用KBr做支撐片,在400~4 000 cm-1范圍內測量紅外吸收峰。使用美國公司Perkin Elmer Diamond TG/DTA熱重分析儀進行熱分析(樣品測試條件:氮氣氛圍下,加熱速率為10℃·min-1,氣流速率 200 mL·min-1,溫度范圍為 30~800 ℃)。 使用德國BRUKER公司AXS D8 X射線衍射儀 (樣品測試條件:Cu Kα,λ=0.154 nm,電壓 35 kV,電流 20 mA,2θ掃描范圍為 5°~90°,步長 0.02°)對制備的樣品進行表征。在上海大學測試中心使用高場振動樣品磁強計 (LS7307-939)設定在27℃測量磁化強度。使用配備有使用微聚焦Al Kα線(1 486.6 eV)單色化X射線源和半球形電子分析儀的Thermo Scientific Escalab 250Xi光譜儀獲得X射線光電子能譜(XPS)。
1.2 制備與測試
1.2.1 氧化石墨烯的制備
參照文獻[10-11]制備氧化石墨烯將69 g的濃硫酸(質量分數98%)倒入500 mL三口瓶中,冰浴15 min,將2.0 g石墨粉緩慢加入到濃硫酸中,冰浴30 min,繼續緩慢加入8.0 g高錳酸鉀,加完后將裝置放入35℃水浴中,攪拌12 h。攪拌后緩慢加6次水,每次46 mL,在35℃條件下攪拌2 h。然后緩慢加入25 mL 30%的H2O2繼續攪拌15 min,反應暫停。把黃色溶液倒進離心管中用離心機離心5 min,轉速8 000 r·min-1。將得到的黃色粘稠物用稀鹽酸(150 mL鹽酸與900 mL水配制)洗滌6次,最后將流動性較強的黃色粘稠物均勻涂在玻璃板上,70℃下烘干。
注意:加高錳酸鉀必須緩慢!用時1~2 h。
1.2.2 乙二胺修飾氧化石墨烯
量取 400 mL的乙二胺,加入到1 000 mL三口燒瓶中。在氮氣氛圍中加350 mg的鋰(碎塊),待鋰完全溶解后,升高溫度(<30℃)使溶液逐漸變為深藍色,攪拌至溶液由深藍色變為灰白色。向反應體系中加入400 mg自制的GO,升溫至50℃攪拌12 h。加入75 mL甲醇和250 mL水,將反應懸浮液過濾,乙醇和水各洗3次,60℃下真空干燥12 h。得到乙二胺功能化的GO[12]。
實驗過程中發現如果先加鋰,使鋰在乙二胺中溶解,在升溫的過程中溶液的顏色由深藍色變為灰白色,然后加石墨烯,可以得到穩定的石墨烯懸浮液。造成上述現象的原因是鋰首先與乙二胺發生反應,生成單取代的鋰胺衍生物作為親核試劑進攻石墨烯的C=C,生成乙二胺功能化的石墨烯。為了消除石墨烯物理吸附乙二胺的影響,用過量的醇和蒸餾水反復洗產物。
1.2.3 乙二胺和丁二胺混溶修飾氧化石墨烯
先將丁二胺(m.p.27~28℃)熔化為液體。用量筒量取120 mL的乙二胺和30 mL的丁二胺,加入到500 mL的三口燒瓶中。在氮氣氛圍中加130 mg的鋰,待鋰完全溶解后,升高溫度(<30℃)使溶液逐漸變為深藍色。攪拌至溶液由深藍色逐漸變為灰白色。向反應體系中加入150 mg自制的GO,升溫至 50℃,攪拌12 h,之后加28.2 mL甲醇和 93.8 mL水。將反應懸浮液過濾,乙醇和水各洗3次,60℃下真空干燥12 h。得到乙二胺和丁二胺混溶功能化的GO。 依次再制備 V乙二胺∶V丁二胺=2∶1、3∶1、5∶1、6∶1 的混溶功能化GO。
1.2.4 乙二胺和己二胺混溶修飾氧化石墨烯
先將己二胺(m.p.39~43℃)熔化為液體。量取160 mL的乙二胺和40 mL的丁二胺,加入到500 mL三口燒瓶中。在氮氣氛圍中加200 mg的鋰,待鋰完全溶解后,升高溫度(<30℃)使溶液逐漸變為深藍色,攪拌至溶液由深藍色逐漸變為灰白色。向反應體系中加入200 mg自制的GO,升溫至50℃,攪拌12 h。之后加37.5 mL甲醇和125 mL水。將反應懸浮液過濾,乙醇和水各洗3次,60℃下真空干燥12 h。得到乙二胺和己二胺混溶功能化的GO。依次制備V乙二胺∶V己二胺=2∶1、3∶1、5∶1、6∶1 的混溶功能化 GO。
1.2.5 四氧化三鐵的制備
稱量 1.800 3 g FeCl3·6H2O,2.002 5 g 乙酸鈉,1.006 1 g聚乙烯吡咯烷酮(PVP)溶解在50 mL乙二醇中,攪拌成均勻透明澄清液體。然后將液體轉移至高壓釜中,200℃下反應8 h。倒出反應釜中的液體,磁鐵分離,乙醇洗3次,60℃下真空干燥[13-15]。
1.2.6 復合物的制備
稱量0.50 g氨基化GO分散在40 mL二甲基甲酰胺(DMF)中,稱量0.40 g Fe3O4分散在60 mL氯仿中,攪拌、超聲使其形成均勻分散液。4 h后將復合物用無水甲醇洗滌3次,磁鐵分離,60℃下真空干燥。將上述的復合物進行編號:乙二胺功能化的GO/Fe3O4磁性復合材料(E),乙二胺和丁二胺混溶功能化的 GO/Fe3O4磁性復合材料(體積比為 2∶1、3∶1、4∶1、5∶1 和 6∶1 分別標記為 B1、B2、B3、B4 和 B5),乙二胺與己二胺混溶功能化的GO/Fe3O4磁性復合材料(體積比為 2∶1、3∶1、4∶1、5∶1 和 6∶1 分別標記為 H1、H2、H3、H4 和 H5)。
1.2.7 時間對吸附染料質量濃度的影響
稱量50 mg結晶紫置入100 mL棕色容量瓶中,定容、超聲。將制備的有機胺功能化的GO/Fe3O4復合材料各稱量20 mg,倒入50 mL離心管中,將配制好的結晶紫染料取25 mL倒入離心管中,恒溫振蕩。每隔1 h取一次液體,磁鐵分離,取上層澄清液體用紫外光譜測試吸附染料的質量濃度。
1.2.8 染料的質量濃度對吸附染料濃度的影響
選取編號為E、B4和H4這3個樣品各稱量10 mg放入50 mL離心管中。配制質量濃度為100、200、300、400、500 mg·L-1的結晶紫染料, 取 10 mL入離心管中,恒溫振蕩。過夜,磁鐵分離,取上層澄清液體用紫外光譜測試吸附染料的質量濃度。
2 結果與討論
2.1 TEM和SEM分析
圖1 GO、Fe3O4和復合材料E的TEM圖。從圖1a可以看出,GO是片狀的,具有多層結構,表面光滑,片層明顯起皺。層與層之間的相互折疊和GO極薄的平面結構產生較大的比表面積,這有利于提高吸附能力。由于GO的長度相對較長并且具有僅幾納米薄的厚度,所以石墨烯納米薄片在溶劑中表現出最低的能量并彼此折疊。在TEM照片中可以清楚的看到,石墨烯納米片具有大的對比度的暗線,應是折疊的多層石墨烯氧化物。基于分析,制備的樣品具有GO的形態特征。從圖1b看出,Fe3O4的形狀是球形的,具有均勻的粒度,平均粒徑約為200 nm。從圖1c和1d可以看出,復合材料E中的Fe3O4很好地固定在樣品中薄薄的GO片層上。GO片層不僅可以防止Fe3O4微球的團聚,而且可以很好地分散這些氧化物微球,并且還可以顯著增強復合材料的比表面積,提高復合材料的吸附性能。

圖1 GO(a)、Fe3O4(b)和復合材料 E(c~d)的 TEM 圖Fig.1 TEM images of GO(a),Fe3O4(b)and composites E(c~d)
圖2 為復合材料E、B4和H4的SEM圖。從圖2中可以看出,復合材料H4(圖2e,2f)要比復合材料E(圖2a,2b)、B4(圖2c,2d)更好地分隔 GO 片層的結構,可以把Fe3O4很好地包裹起來,而Fe3O4也很好地穿插在GO片層上。這是由于己二胺的分子鏈長比丁二胺的長,而丁二胺的又比乙二胺的長,所以H4中的己二胺可使GO的結構更易分層。另外,由于Fe3O4具有磁性,所以圖中的Fe3O4有團聚現象。

圖2 復合材料 E(a~b)、B4(c~d)和 H4(e~f)的 SEM 圖Fig.2 SEM images of composites E(a~b),B4(c~d)and H4(e~f)
2.2 紅外光譜圖
復合材料的FT-IR光譜如圖3所示。在557 cm-1處產生的強吸收帶是Fe-O鍵,可以看出Fe3O4成功附著在氨基化GO表面。在3 433 cm-1處產生的強而寬的吸收峰是-NH2的伸縮振動。在1 076 cm-1處對應于C-O鍵的伸縮振動,在1 400 cm-1處附近則是-OH的面內變形振動峰,峰的強度較弱,峰形較寬。在1 629 cm-1處為石墨中sp2雜化C=C雙鍵伸縮振動[16],而2 926 cm-1處則為C-H吸收峰不對稱伸縮振動。紅外光譜數據表明,所制樣品為有機胺功能化的GO/Fe3O4復合材料。
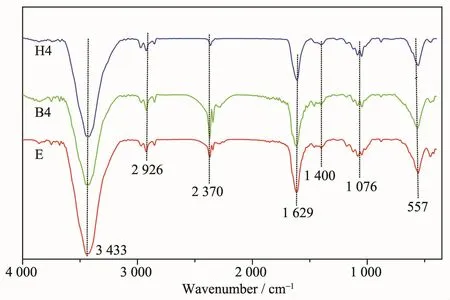
圖3 復合材料E、B4和H4的紅外譜圖Fig.3 FT-IR spectra of composites E,B4 and H4
2.3 熱重分析(TG)

圖4 GO與復合材料E的TG曲線Fig.4 TG curves of GO and composites E
GO與復合材料E的熱重曲線如圖4所示(復合材料B4和H4的失重與E大致相同,故未列出)。可以看出,GO與復合材料E的失重大致分為3個階段。第一階段,GO在20~187℃大約有17.2%的失重,而復合材料E在20~145℃大約有2.2%的失重,對應于GO中存在有的少量水和乙醇 (沸點為78.3℃)等,包括物理吸附和化學鍵合水,表明GO有很強的親水性。第二階段,GO在130~306℃大約有42.2%的失重,是由于GO表面上的含氧官能團(如-OH,-COOH,-O-和C=O)受熱分解導致。而復合材料在145~922℃大約有32.6%的失重,這是GO表面上的含氧官能團和C-C鍵碳骨架在依次受熱分解。第三階段,GO在306~983℃失重大約有13.0%,這就是GO上C-C鍵碳骨架在受熱分解。復合物在922~983℃大約有15.4%的失重,這可能是氨基在受熱分解。復合物在1 000℃的時候質量分數要比氧化石墨烯的質量分數多出約22.2%,這應該是Fe3O4(熔點為 1 594 ℃)。
2.4 X射線衍射(XRD)圖和磁滯回線
如圖5a所示,復合材料E的衍射峰出現在18.47°,30.32°,35.67°,43.24°,53.62°,57.16°,62.82°,分別對應于立方相 Fe3O4的(111),(220),(311),(400),(422),(511),(440)晶面,與標準衍射卡 PDF#19-0629上的衍射峰一致[15]。GO在2θ=10.16°處產生了尖銳的衍射峰,對應于晶面(001)[17]。而有機胺功能化的GO/Fe3O4復合材料B4、H4的衍射峰大致與Fe3O4的相同,GO的衍射峰不太明顯。有機胺的加入未使復合物產生相變。測定的復合材料E的磁滯回線如圖5b所示。其磁滯回線為S型曲線,故E是超順磁性的,比飽和磁化強度 Ms為 53.0 emu·g-1。
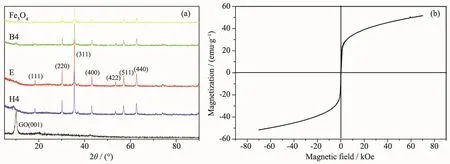
圖5 (a)GO、復合材料E,B4、H4以及Fe3O4的XRD圖;(b)復合材料E的磁滯回線圖Fig.5 (a)XRD patterns of GO,composites E,B4,H4 and Fe3O4;(b)VSM of composites E
2.5 X射線光電子能譜(XPS)分析
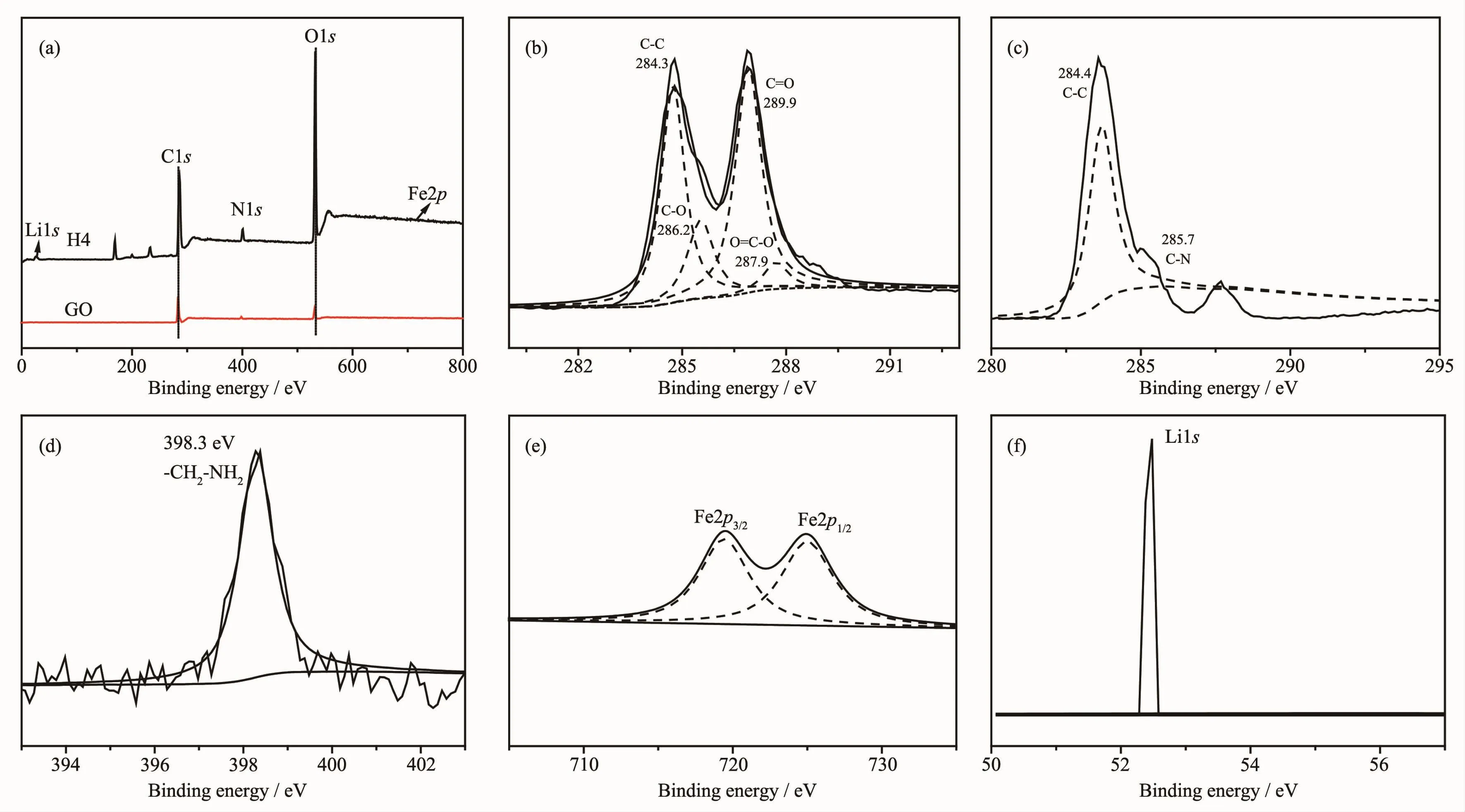
圖6 GO 和 H4(a),GO C1s(b),H4 C1s(c),H4 N1s(d),H4 Fe2p(e)和 Li1s(f)的 XPS 譜Fig.6 XPS spectra of GO and H4(a),GO C1s(b),H4 C1s(c),H4 N1s(d),H4 Fe2p(e)and Li1s(f)
XPS分析可用于確定改性所引入的化學物質。圖6a中,在285.6、400.6和531.9 eV處的峰分別對應于 C、N 和 O[18],在 52.5 eV 處的峰為 Li[19],位于719.4 eV峰為Fe[20]。圖6b中,位于284.4 eV的GO的C1s峰其結合能位于284.3、286.2、286.9和 288.4 eV,它們分別對應 C-C,C-O,C=O 和 O=C-O[20]。 圖6c描述了H4的C1s,其中C-C鍵的主峰位于284.4 eV,胺(CH2-NH2)中的C-N鍵的第二個峰位于285.7 eV[21]。從圖6d可以看出,N1s結合能位于398.4 eV,對應于-CH2-NH2[22]。圖6e證明了Fe的組成[20,23]。圖6f中位于52.5 eV的結合能為Li1s[19]。
2.6 時間對吸附染料質量濃度的影響
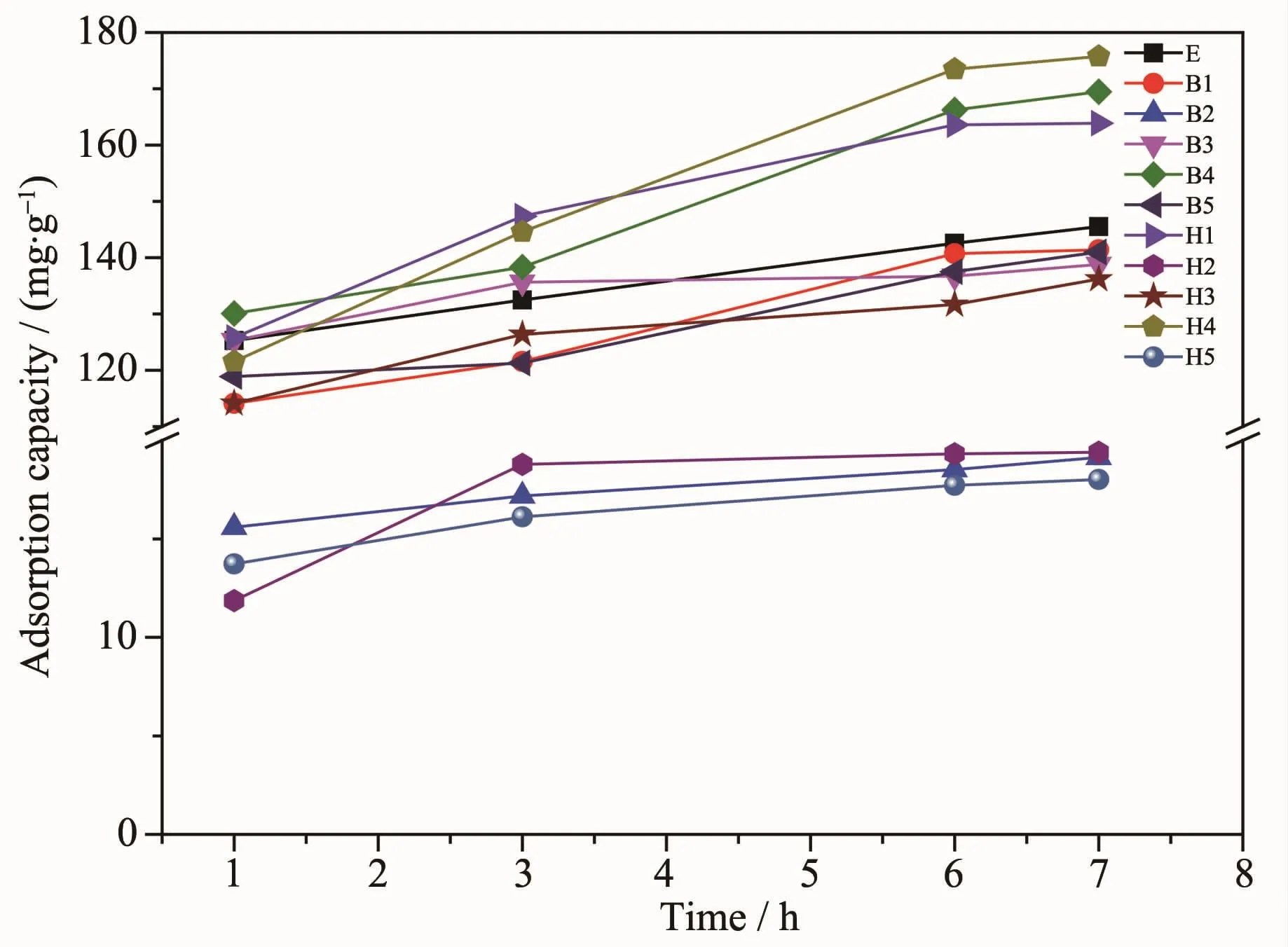
圖7 不同復合材料對結晶紫最大吸附量隨時間的變化Fig.7 Change of maximum adsorption capacity of crystal violet on different composites with time
圖7 表示不同藥品對結晶紫染料最大吸附量隨時間的增加而增加,結晶紫染料濃度為500 mg·L-1。從圖7中可以看出,有機胺功能化的GO/Fe3O4磁性復合材料在吸附結晶紫染料時,隨著時間的增加,復合材料對結晶紫的吸附量越大。由實驗可得6 h后最大吸附量基本不再變化。從圖7中可以看出,乙二胺與己二胺混溶功能化的GO/Fe3O4磁性復合材料H4對結晶紫染料的吸附量最大,為173.2 mg·g-1。
2.7 染料濃度對吸附染料質量濃度的影響
根據時間對吸附結晶紫染料的影響,選取復合材料E、B4和H4。從圖8中可以看出,復合材料對結晶紫染料的最大吸附量隨質量濃度增大而增大,而移除率卻隨結晶紫染料質量濃度增大而減小,最后都趨于定值。由這3種復合材料比較得出復合材料H4吸附結晶紫染料的量最多,B4其次,這驗證了己二胺要比乙二胺和丁二胺的分子鏈要長,它改性GO要比乙二胺和丁二胺這2種藥品對GO改性分層隔離得要開,比表面積變大吸附性能增強。結晶紫染料的移除率可表示為:
E=(C0-Ce)/C0×100%
其中,E為移除率,C0和Ce分別為結晶紫染料的初始質量濃度和吸附處理后的質量濃度。復合材料H4在濃度為 400 mg·L-1時最大吸附量為 164.3 mg·L-1,濃度為100 mg·L-1時最大移除率為68.1%。
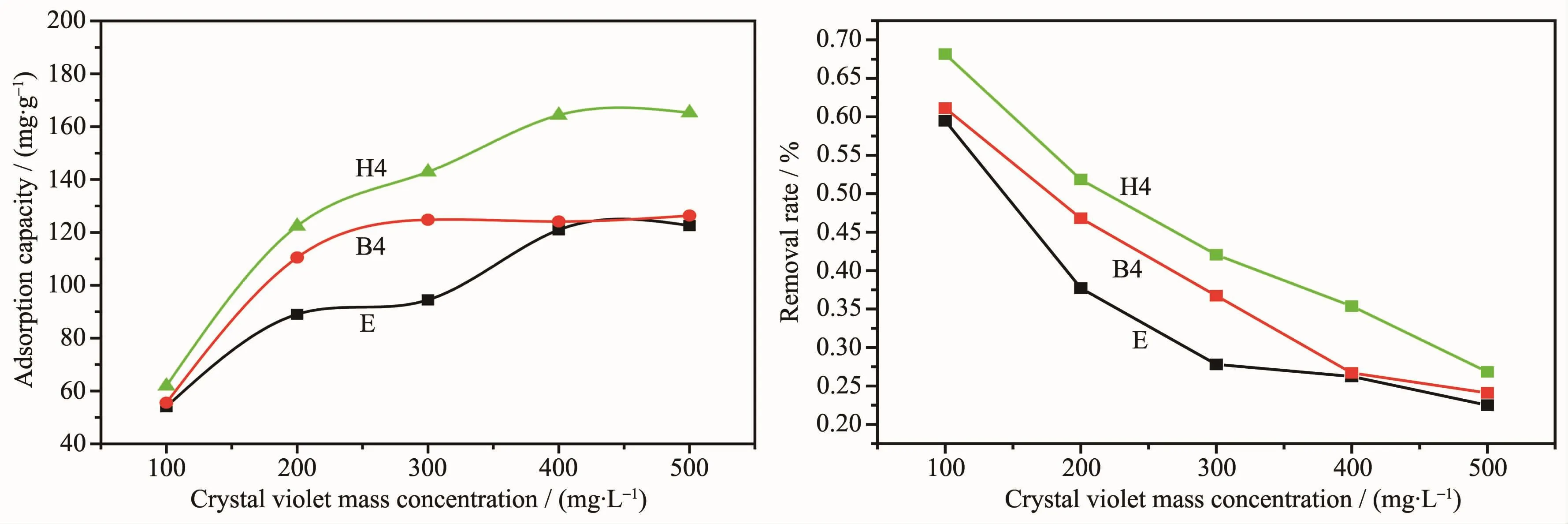
圖8 復合材料E、B4、H4吸附結晶紫的最大吸附量和移除率Fig.8 Maximum adsorption amount and the removal rate of the crystal violet on composites E,B4 and H4
3 結 論
用乙二胺、乙二胺與丁二胺/己二胺混溶來改性氧化石墨烯,并用物理混合法制備了GO/Fe3O4/有機胺的三元復合體系。復合物中,GO為片層結構,Fe3O4粒徑分布均勻,可以很好地穿插在氨基化GO片層上,由于己二胺的鏈長更長,它要比乙二胺和丁二胺更好地隔離GO片層。
磁性復合材料對結晶紫染料具有吸附能力,且能夠借助Fe3O4的磁性,實現快速分離。乙二胺和己二胺混溶功能化的GO/Fe3O4磁性復合材料要比其它2種胺改性的復合材料吸附更好,對結晶紫的最大吸附量隨染料的濃度增大而增大,隨時間增長而增大,最后都趨于定值。 其中,V乙二胺∶V己二胺=5∶1 時制得的復合材料H4吸附性能最好。
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
