基于材料基因組方法的鋰電池新材料開發?
2018-07-10 09:32:48肖睿娟李泓陳立泉
物理學報 2018年12期
肖睿娟 李泓 陳立泉
(中國科學院物理研究所,清潔能源重點實驗室,北京 100190)
(2018年4月11日收到;2018年4月27日收到修改稿)
1 引 言
可充放鋰二次電池是下一代高能量密度電池最有希望的電池體系,它是電動汽車技術成敗的關鍵,也是風電和光伏電儲能的首選[1].能量密度高、安全性好、充放電速度快的下一代鋰二次電池是主要的發展方向[2].在鋰二次電池技術的發展歷程中,每一次電池性能的顯著提升都與新材料的發現和發明息息相關[3].從廣泛應用于消費電子產品的鈷酸鋰LiCoO2正極到摻雜改性的三元LiNixCo1?xMnxO2正極材料,有效提高了鋰離子電池的能量密度;磷酸鐵鋰LiFePO4則以其穩定性而成為高安全性的正極材料;硅負極材料經歷從設計到產業化的歷程,成為下一代高容量負極的首選材料;鈦酸鋰則由于其“零應變”的特性成為具有高循環穩定性的負極材料.可以預見,新材料的發現仍然是促進下一代鋰二次電池發展的關鍵.
傳統的電池材料研發是基于以“試錯法”為特征的開發模式,從發現到應用的周期很長,一般需要20年或更長時間.為加速材料從研究到應用的進程,美國政府于2011年6月提出了“材料基因組計劃 (Materials Genome Initiative)”,希望通過材料研發模式的革新,將材料從發現到應用的速度提高一倍,成本降低一半[4].目前,美國、日本和歐盟等發達國家均作為國家戰略支持了一批科研機構開展將“材料基因組”思想應用于鋰電池材料開發的研究.我國也及時在該方向開始了探索.“材料基因組”科學研究的關鍵是實現材料研發的“高通量”,即并發式完成“一批”而非“一個”材料樣品的計算模擬、制備和表征,即高通量計算、高通量制備與高通量表征,實現系統的篩選和優化材料,從而加快材料從發現到應用的過程.利用“材料基因工程”方法,通過高通量、多尺度的大范圍計算和搜索,借助數據挖掘技術和方法,有望篩選出可能具有優異性能的新材料,從而探索材料基因組方法在研究開發鋰電池材料和先進功能材料中的應用.本文針對鋰二次電池材料中特有的科學基礎問題,介紹材料基因組方法的發展及其對下一代鋰二次電池關鍵材料開發的促進作用.
2 材料基因組方法的發展與探索
2.1 鋰二次電池材料研究中的基礎科學問題
在鋰二次電池的工作過程中,電池材料中電子、離子、界面的性質與電池器件的能量密度、安全性、充放電速率、循環穩定性等性能有著密不可分的聯系.如圖1所示,電池在充放電循環過程中的電荷轉移、電子輸運、電荷分離等現象與電極材料中的電子結構相關;而電極與電解質材料中的離子傳輸性質則決定了Li+在電池工作過程中的擴散路徑、擴散勢壘和擴散系數,與電池的充放電速率、極化現象等相關;電極/電解質界面處的微觀結構則與循環過程中的熱穩定性、電化學穩定性相關聯,與電池器件的循環性能密不可分[5].因此,要發展更高能量密度、更高安全性和更快充放電速率的鋰二次電池,需要針對與各個目標相關的材料性質進行改進.例如:通過提高電極材料的可轉移電子數和可轉移離子數來提高其儲鋰容量,獲得更高能量密度的正、負極材料;通過探索與鋰離子運動快慢相關的結構因素,找尋離子電導率更高的電解質材料,獲得更快的電池充放電速率;通過研發不含有機可燃電解質、并具有穩定電極/電解質界面的固態電池來改善器件整體的安全性等[1,2].
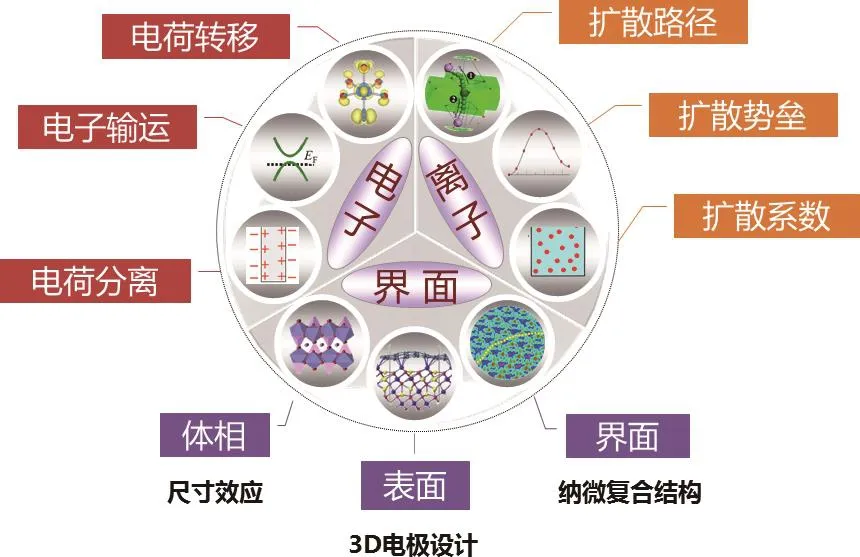
圖1 鋰二次電池器件中存在的基礎科學問題Fig.1. The basic scienti fi c questions existing in lithium secondary batteries.
隨著電池研究的不斷發展,全固態電池已成為下一代鋰二次電池的主要研究方向(圖2).在采用固體電解質替換傳統使用的有機液態電解質后,有望避免有機電解質的揮發、分解、可燃等隱患,從而提高電池的安全性[6?8].此外,由于采用了固態電解質,使得金屬鋰負極的應用成為可能,這就為正極材料的選擇提供了更為廣闊的空間,使得不含鋰的化合物也有可能用作鋰電池的正極.在鋰二次電池的中期研究目標中,采用金屬鋰負極、固體電解質和高容量正極材料的全固態電池可獲得300—400 W·h/kg的能量密度.然而,鋰離子在固體中的傳輸速度通常比其在液態中的傳輸速度慢1—2個數量級,因此,發現具有高離子傳輸速度的固體電解質十分重要[9].同時,原本傳統電池中電極與液態電解質之間的固/液界面在全固態鋰電池中成為固/固界面,隨著鋰離子在電極材料中的脫出和嵌入,電極材料的體積會發生變化,此時,電極與電解質之間固/固界面的力學穩定性、熱力學穩定性和電化學穩定性等都成為影響電池循環壽命的重要因素,發現鋰脫嵌時體積變化小的“低應變”材料可提高界面的穩定性[10,11].由此可見,發明具有高離子電導率的固態電解質與體積變化小的低應變電極是全固態鋰電池材料發展的關鍵.
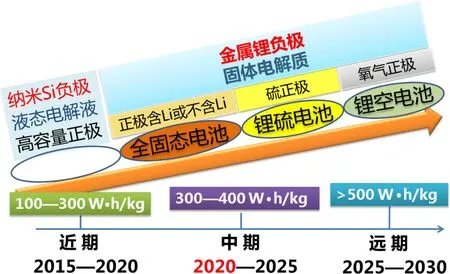
圖2 下一代鋰二次電池器件的發展路線圖Fig.2.The roadmap for the next-generation lithium secondary batteries.
2.2 適用于鋰電池材料開發的高通量計算方法發展
鋰二次電池材料中,鋰離子的輸運快慢與電池性能息息相關.然而,基于量子力學方法的離子輸運性質計算的運算量非常大,每種結構的計算常常耗時幾天.因此,基于離子輸運性質的量子力學計算不適合于直接用來發展高通量算法,需要通過合理設計篩選流程和發展適合于離子輸運性質篩選的計算方法來實現對關鍵材料固體電解質的開發[12].
離子輸運性質的計算方法可分為圖3所示的三種:根據鋰離子是在晶格中一定的幾何空間里運動,因此可通過Voroni-Dirichlet等空間分析方法來判斷晶格中是否存在鋰離子可運動的通道[13];更進一步的方法考慮了鋰離子在晶格中的成鍵情況,根據鋰離子在晶格中鍵價失配度來判斷鋰離子的通道和勢壘值,這種方法基于半經驗勢函數,計算量小,對單個晶體結構僅需幾分鐘就可完成計算[14,15];最為精確的計算模擬方法是采用基于密度泛函理論(DFT)的量子力學計算,結合過渡態理論或分子動力學方法,得到能量勢壘低的遷移路徑,這種方法計算精度高,但運算量大,對于單個離子遷移路徑,通常也需要若干小時至若干天的運算時間[16].顯然,運算量過大是限制計算方法高通量化的主要原因.

圖3 計算離子輸運性質的幾類方法及對應的計算量Fig.3.The calculation methods for ion conduction and the computation costs.
針對各種計算方法的特點,我們設計了將不同精度計算方法相結合的高通量篩選流程[17].基于化學成鍵作用的鍵價計算程序可在數分鐘內半定量地模擬出材料中的鋰離子輸運路徑和遷移勢壘,非常適合于大范圍計算的高通量篩選.基于DFT的過渡態計算能準確地計算出離子輸運的勢壘,但計算量大、耗時長,適合于對單個材料進行深入研究.基于此,我們將不同精度的計算方法用于材料計算篩選的不同階段:首先依據材料的使用條件通過元素篩選縮小范圍,然后采用快速的鍵價計算進行初步篩選去除離子輸運勢壘較大的化合物,最后采用基于密度泛函的模擬對上一步篩選得到的材料進一步精確計算獲得最終的備選材料,從而有效地提高了整體的篩選效率,實現了鋰二次電池材料中快離子導體的高效篩選.
基于上述篩選思路,我們采用模型材料計算了Li+遷移時在庫侖場中的庫侖能變化,通過與DFT計算得到的遷移勢壘相比,發現了兩者之間的對應關系,由此說明了庫侖作用與離子遷移勢壘的相關性.在此基礎上,通過引入Adams和Prasada Rao[15]改進的鍵價和理論來描述鋰離子與陰離子之間的成鍵作用,由此開發出了能快速地定性預測材料中離子輸運路徑和遷移勢壘的計算程序BV-path[17].通過編寫圖4中的一系列腳本實現了從無機晶體結構數據庫中的晶體結構文件自動生成計算所需要的結構、占位、電荷等輸入文件,并自動提交運算,進行超晶胞建立、空間格點生成、鍵價和計算、勢能計算等過程,最后自動輸出計算結果,獲取晶體結構中可能存在的離子輸運路徑與遷移勢壘.我們采用高通量的自動化計算方法,對無機晶體結構數據庫中1000多種含鋰的氧化物進行了鋰離子輸運路徑和遷移勢壘的計算,獲得了一系列化合物的離子輸運性質數據[12].通過將計算結果與采用密度泛函過渡態理論計算得到的勢壘數據相比較,證實了采用半經驗勢的鍵價方法能有效地給出不同晶體結構中離子輸運性質的難易程度,其預測結果可給出與密度泛函相同的變化趨勢,可定性地描述離子在晶體中的傳輸過程,從而可以有效地用于離子輸運性質的初步篩選.
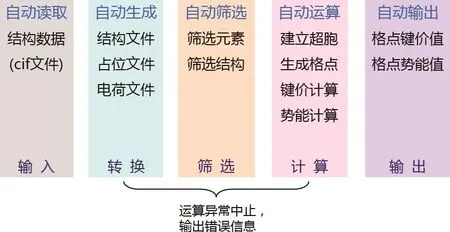
圖4 通過一系列命令腳本實現運算過程的自動化Fig.4. The series of scripts to realize the highthroughput calculation automatically.
3 高通量計算方法應用于鋰二次電池的新材料設計
3.1 富鋰正極新型包覆材料的篩選
在全固態電池中,電極/電解質界面的穩定性和離子電導率都影響著電池的整體性能.在電極表面包覆上能傳輸鋰離子并阻隔電極、電解質之間發生反應的化合物往往能改善電池的綜合性能.若包覆物與電極具有相似的晶體結構,將更易于形成結合緊密、應力較小的界面層.從高通量計算篩選的1000多種含鋰氧化物中,不僅能發現離子輸運勢壘較低的快離子導體備選材料[12],而且還發現了一些與目前所研究的電極材料具有相似結構并且能傳導鋰離子的化合物,這些化合物有可能用作電極的包覆材料,從而改善電極的綜合性能.
基于材料基因組思想,通過采用高通量計算篩選,綜合考慮結構匹配、擴散通道、導電性等因素,發現了兩種可能與鋰離子電池富鋰正極材料相匹配的包覆化合物Li2SiO3和Li2SnO3[18].這兩種材料都屬于離子化合物,具有較好的離子導電性,并且在化學結構上與富鋰材料((1?x)Li2MnO3·xLiMO2)中的母相材料Li2MnO3相似,因此可嘗試選擇其作為富鋰材料的表面修飾層.圖5展示了Li2SiO3和Li2SnO3的晶體結構及采用鍵價方法計算得到的離子輸運通道,可以看出它們具有與Li2MnO3[17]相似的層狀結構及二維離子輸運通道.
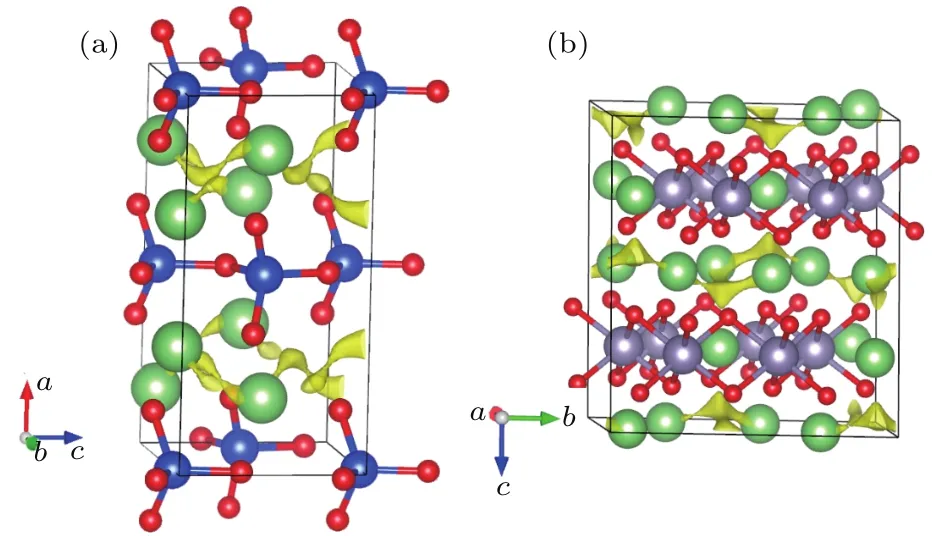
圖5 用鍵價方法計算得到的(a)Li2SiO3和(b)Li2SnO3的離子輸運通道Fig.5.The Li+migration pathways in(a)Li2SiO3and(b)Li2SnO3simulated by bond-valence method.
通過實驗制備包覆后的富鋰正極材料并測試其電化學性能,證實了包覆材料Li2SiO3和Li2SnO3可有效改善Li2MnO3電極的循環穩定性[18].對比包覆前后電極的循環伏安曲線、交流阻抗譜等實驗結果,發現包覆的主要作用是加速了電極擴散速度,抑制了循環過程中界面阻抗的增大,從而改善了電極的反應動力學.其中Li2SiO3修飾后的材料在表面會與電解液發生化學反應,生成路易斯酸.生成的這種強酸一方面能夠加速過渡金屬離子的溶出,中和材料表面的堿性絕緣雜質,另一方面形成的表面固溶體有利于表面鋰離子的擴散,使材料表現出更好的電化學性能和結構穩定性.而Li2SnO3為修飾層的材料在活性材料表面作為保護層不僅能夠保護材料免受電解液的腐蝕,抑制過渡金屬離子的溶出,而且提高了材料的動力學行為,從而使材料具有較好的電化學性能.
3.2 高通量計算篩選固體電解質β-Li3PS4的優化改性方案
目前從改善鋰二次電池安全性的角度考慮,全固態鋰電池被公認為未來二次電池的重要發展方向.在傳統的鋰二次電池中,電解液體系采用了液態可燃的有機溶劑,而全固態鋰二次電池則采用穩定的固態電解質材料,有望從根源上改善電池的安全性問題.此外,在全固態鋰二次電池中,有可能采用金屬鋰作為負極材料,而金屬鋰負極的理論容量是鋰電池體系的最高極限,其電位也是所有負極材料中最低的,因此可以大大提高鋰二次電池體系的能量密度.然而,使用固體電解質材料體系的一個最大問題是鋰離子在固體電解質材料中的離子輸運很慢.一般來說,鋰離子電導率在固體電解質中要比在液體電解液中低1—2個數量級.開發兼具高離子電導率、高穩定性、高機械強度的固體電解質材料勢在必行.
在已經研究過的固態電解質材料中,硫化物作為目前發現的具有最高鋰離子電導率的固體化合物,一直是研究中的熱點[19].然而硫化物在空氣中的敏感性以及它在與氧化物正極界面處表現出的化學不穩定性,極大地限制了硫化物在固態鋰電池中的實際應用[20,21].Li-P-S體系是目前發現的穩定性最高的含鋰硫化物體系,對該體系中的組分進行改性優化,有望在提高其離子電導率的同時進一步提升其穩定性,從而獲得新的固態電解質備選材料.通過采用密度泛函計算與鍵價計算相結合的方法,可以對大量的摻雜改性方案進行高通量的計算篩選.采用可準確確定晶體結構的密度泛函計算來獲得摻雜后的原子位置信息,再通過鍵價計算快速選擇其中有利于降低鋰離子遷移勢壘的摻雜方案.通過對β-Li3PS4的P位進行Sb,Zn,Al,Ga,Si,Ge,Sn的摻雜,以及對S位進行O摻雜的研究發現,用氧替換晶格中部分硫或用鋅氧兩種元素對β-Li3PS4進行共摻雜能有效提高其離子電導率(圖6)[17].進一步關于其穩定性的計算表明,氧的摻入提高了β相的穩定性,降低了該材料在室溫附近向γ相轉變的趨勢[17,22].
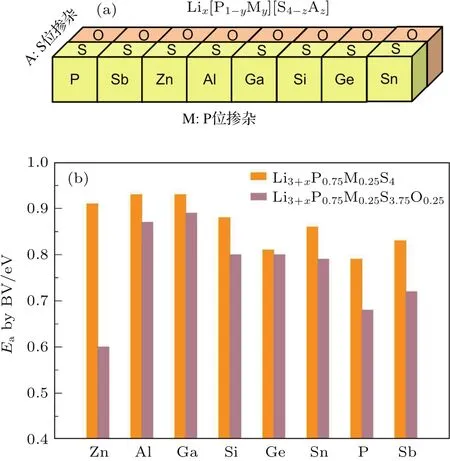
圖6 (a)采用密度泛函計算與鍵價計算結合的高通量計算流程,篩選能改善β-Li3PS4離子電導率和穩定性的摻雜改性方案;(b)P位摻雜Sb,Zn,Al,Ga,Si,Ge,Sn以及S位摻雜O后計算得到的鋰離子遷移勢壘[17]Fig.6.(a)High-throughput screening for the doping strategy of β-Li3PS4by the combination of density functional theory and bond-valence(BV)calculations;(b)calculated Li+migration energy barriers for Sb,Zn,Al,Ga,Si,Ge,Sn doped P-site and O doped S-site[17].
在通過高通量計算篩選獲得了材料改性的優化方案后,基于密度泛函的高精度計算可有效揭示摻雜對材料性能的改善機理.計算得到的電子結構表明上述性能的改善來源于P—O鍵與P—S鍵之間的差異.氧摻雜β-Li3PS4引起了[PS4]3?單元局域結構變化,在摻雜單元[PS3O]3?附近提供了更多供鋰離子運動的空間,形成了新的離子輸運路徑,使得原本的二維輸運方式轉變為三維輸運方式.對β-Li3PS4與金屬鋰界面的模擬顯示,與鋰接觸的[PS3O]3?單元雖然發生顯著的形變,但并未發生化學鍵的斷裂,從而穩定了兩者的接觸界面[23].
3.3 高通量結構預測方法發現全新結構的固體電解質LiAlSO
在通過高通量計算篩選固體電解質β-Li3PS4優化改性方案的過程中,氧摻雜顯示出了對硫化物穩定性提升的顯著效應.在目前探索的固體電解質材料中,氧化物和硫化物均顯示出各自的優勢.與硫化物相比,氧化物具有更高的穩定性,也更加容易制備,然而其離子電導率仍然遠低于硫化物.而硫化物雖然具有較高的離子電導率,但使其穩定存在的條件更為苛刻,且制備過程更為困難.考慮到硫化物和氧化物各自的特點,我們提出了設計多種陰離子共存的快離子導體設計思想,并嘗試設計含鋰的氧硫化物,以集成硫化物高離子電導率和氧化物高穩定性的優點.
高通量結構預測算法可用于在已知元素組成的空間里設計全新的晶體結構,該方法已成功應用于高壓條件下新型化合物的尋找[24?26].通過采用CALYPSO軟件[27]在Li-Al-S-O的元素空間中構建具有各種空間群的晶體結構,并對其進行結構優化和能量計算,基于其中能量低的結構運用粒子群優化算法生成新的結構,在此優化過程中,逐漸找到由這四種元素按照1:1:1:1的比例形成的最穩定結構.計算結果顯示[28],這種全新的氧硫化物LiAlSO具有與β-NaFeO2相似的正交結構,AlS2O2層沿b軸方向平行排列,Li離子位于層間與S和O形成扭曲的四面體單元(圖7).基于密度泛函的計算表明,Li離子在該材料中沿a軸方向的離子遷移勢壘低于50 meV,是典型的快離子導體,因此,該化合物作為一種全新的快離子導體,可以成為固體電解質的備選材料,并且兼具高離子電導率和改善的化學穩定性.
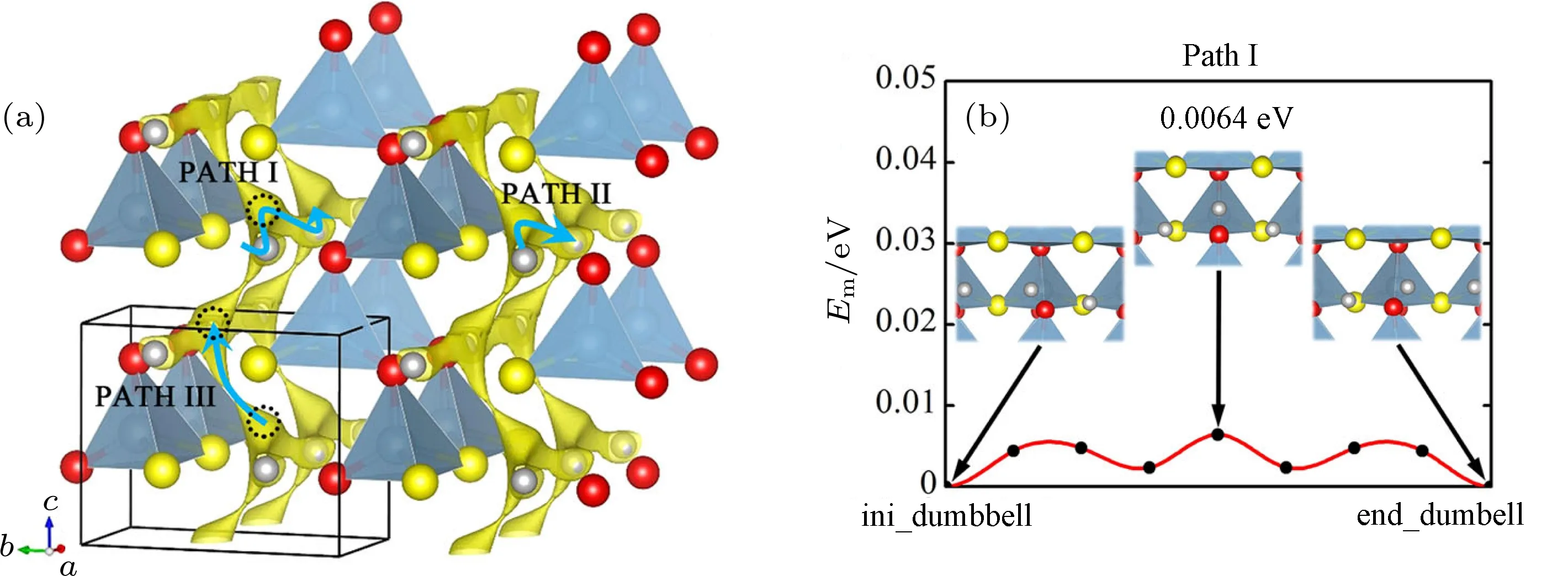
圖7 (a)采用高通量晶體結構預測算法得到的含鋰氧硫化物LiAlSO的晶體結構;(b)密度泛函計算得到的鋰離子在該結構中的輸運勢壘[28]Fig.7.(a)New oxysul fi de LiAlSO designed by high-throughput crystal-structure prediction calculations;(b)the lithium ion migration barriers simulated by using density-functional theory method[28].
3.4 數據挖掘方法研究零應變電極材料中結構與體積變化的關聯
基于材料基因思想的高通量計算與高通量實驗測試為新材料研發領域不僅提供了新的研究思路,而且帶來了成倍增長的數據信息,為大數據方法在材料學中的應用打下了基礎[29?31].在電池材料中還存在許多尚未研究清楚的現象,探尋結構與性能之間的關聯有助于建立起搜尋具有目標物性材料的快捷方法.機器學習技術已被用于獲取材料性質與各種復雜的物理因子之間的統計模型,例如通過預測分子的原子化能尋找熱力學穩定的新化合物[32,33].
在固態鋰二次電池中,固態電解質/電極界面上的穩定性與電池的循環特性緊密相連,其中界面的力學穩定性主要由電極在脫嵌鋰過程中的體積變化決定.電化學循環過程中,固態電解質的體積并不發生顯著變化,而電極材料則在鋰含量增加和減少的過程中會發生體積的變化,若體積變化過大,會造成固態電解質與電極之間接觸界面的松散,影響離子在界面的傳輸[34].因此,尋找脫嵌鋰過程中體積變化小的“低應變”材料十分必要.基于第一性原理計算得到正極材料在脫鋰前后的晶胞體積,以及根據晶體結構、組分、元素、電荷等信息提取描述因子,在此基礎上嘗試采用機器學習中的多元線性回歸分析來尋找影響體積變化的主要因素,構建微觀結構與體積變化之間的關聯[35].
圖8顯示了采用數據挖掘方法研究目標變量與描述因子之間關聯的三個主要步驟.首先需要進行數據準備,獲得不同樣本中目標變量的數據,這里針對尖晶石結構的正極材料LiX2O4和層狀結構的正極材料LiXO2(X為可變價元素)共28種結構,通過密度泛函計算對材料在脫鋰前和完全脫鋰后的結構進行優化,獲得由于脫鋰導致的體積變化百分比.接下來需要對每個樣本建立一系列描述因子,用于表述其原子層面的微觀信息,由于并不清楚哪些因子與體積的變化有關,因此列出了盡量多的描述因子,期望通過數據挖掘模型的構建探尋這些因子與目標物性(體積變化)之間的關聯.在本研究中,為每種結構選取了34個描述因子,包括與晶格參數相關的7個參數、與組成元素基本性質相關的10個參數、與局部晶格形變相關的12個參數、與電荷分布相關的3個參數和與組分相關的2個參數.在具備了描述因子與目標變量的數據后,就可開始采用數據挖掘的方法來建立因子與變量之間的關聯,對于所建立的模型,需要采用統計參數來評估其可靠性及預測能力,并在合理的預測范圍內對新的結構進行目標物性的預測.
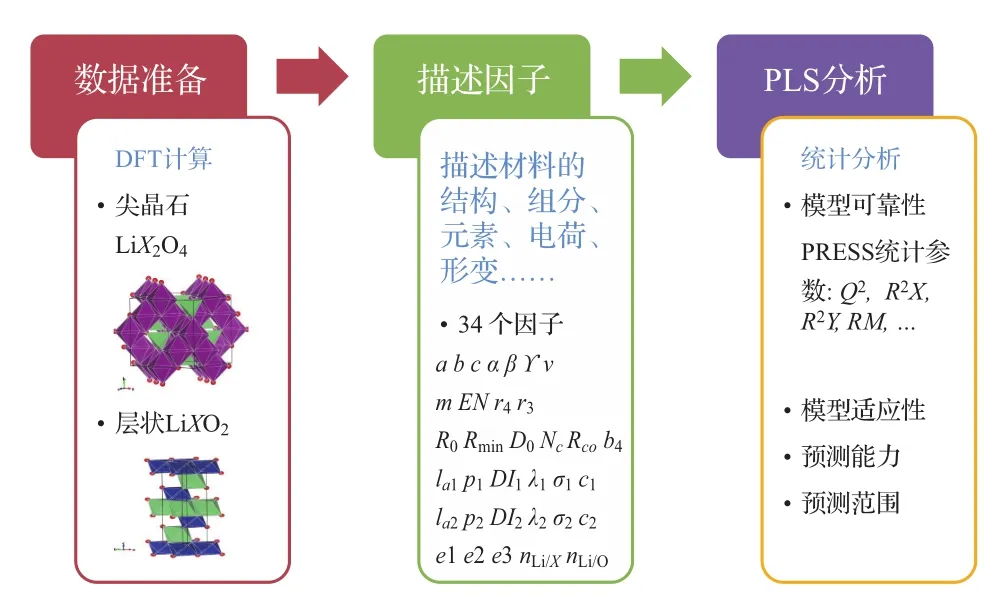
圖8 采用多元線性回歸數據挖掘方法分析脫鋰前后晶格體積變化與結構之間的關聯Fig.8.Three steps for searching the relationship between volume change and atomic structure by partial least squares(PLS)analysis.

圖9 采用PLS模型因子重要性分析探尋對正極材料脫鋰過程體積變化影響較大的參數[35]Fig.9.Variable importance in projection plot of the independent variables for the modelling[35].
通過采用“Leave-One-Out”方法進行評估,發現在上述問題中采用11個相關變量(11 components)時得到的Q2指數最大,表明此時得到的模型最為穩定.進一步的因子重要性分析表明(圖9),盡管離子半徑是晶格體積變化的重要決定因素,但體積變化并不僅僅與離子半徑有關,過渡金屬的成鍵參數及過渡金屬氧八面體的局域結構也對體積變化起到作用.在此模型的基礎上,可以構建含有多種過渡金屬的正極材料,共同調節體系在脫嵌鋰過程中的體積變化,最大程度地減小由于鋰含量變化導致的晶格體積變化率.
4 結 論
針對固態鋰二次電池的研發,我們及時開展了適用于鋰電池材料的高通量計算方法的探索,發展了包含離子輸運性質在內的、融合不同精度的計算方法,建立了基于鋰離子輸運勢壘的高通量計算篩選和優化流程,實現了多種材料的并發計算、監控計算中間過程、分析計算結果、基于計算結果對材料性能的判斷和考核等功能.運用該自主研發的高通量計算平臺,已成功篩選了無機晶體結構數據庫中含鋰的氧化物[12],發現了兩種能改善富鋰正極循環性能的包覆材料;并對硫化物固體電解質進行了摻雜方案的高通量計算優化[17],由此提出了構建多種陰離子共存的固體電解質的設計思想,發明了一種全新的氧硫化物固體電解質[28];根據高通量計算所匯集的數據,嘗試了在正極材料脫鋰過程中的體積變化研究中采用多元線性回歸的數據分析方法[35],為進一步在鋰二次電池研發中引入數據挖掘和機器學習等工智能方法提供了可能.
然而,固態鋰二次電池作為一個復雜系統,單一性能指標的材料篩選和優化過程不能保證尋找到綜合性能優異的電池材料.基于材料基因思想的高通量計算方法將在多目標篩選及材料匹配度篩選方面進一步發展(圖10).以固體電解質為例,其自身至少需要兼具高離子電導率、寬電化學窗口、高穩定性、高機械強度等特點[1,2].目前發現的無機晶態電解質、玻璃態無定形材料體系和聚合物固態電解質材料體系都各有優缺點,顯然僅考慮離子電導率無法獲得綜合性能優異可實際應用的固體電解質材料.因此進一步發展多目標優化的高通量計算篩選流程勢在必行.固態鋰二次電池研發的關鍵是新型固態電解質和高能量密度電極材料的開發,因此有必要發展針對固態電解質的基于鋰離子電導率、電化學窗口、化學穩定性、熱力學穩定性等多目標參數和針對電極材料的基于電極電位、電荷轉移、體積變化、結構穩定性等多目標參數的高通量計算篩選和優化流程.此外,固態鋰二次電池是由正極、固體電解質、負極組成的復雜體系.電池性能不僅與三類材料的本征性質有關,而且受到各部分之間相互作用的制約.要提高固態鋰二次電池的能量密度、循環壽命和安全性等性能,除對單一材料體系進行篩選和優化外,還需要通過高通量計算選擇出合適的電極/電解質組合,使得電極/電解質界面處具有高機械穩定性、低界面阻抗和高電化學穩定性,從而實現對鋰二次電池體系的優化,提高電池體系的整體性能.因此,還需要發展適用于固態鋰二次電池電極/電解質匹配的高通量計算篩選和優化方法以提高電池的綜合性能.
在固態鋰二次電池新材料的高通量計算研發過程中所發展的方法可以方便地推廣到其他類型功能材料的研發中.除了鋰二次電池中關注的離子輸運性質外,基于材料基因思想發展出的技術還可用于磁、電、光、聲、力、熱等多種性質的計算與篩選、優化、預測中.這種新模式下先進功能材料的開發經驗,為材料基因工程方法推廣到更多高新科技發展方向,如包含很多新奇量子現象的超導、量子霍爾效應、量子磁性、磁電耦合等材料,奠定了基礎,將加速各種新材料的發現和發明過程.

圖10 基于材料基因思想的電池材料研發方法的進一步發展趨勢Fig.10.Development of the research method based on materials genome initiative in the near future.
[1]Tarascon J M,Armand M 2001 Nature 414 359
[2]Goodenough J B,Kim Y 2010 Chem.Mater.22 587
[3]Li H,Wang Z X,Chen L Q,Huang X 2009 Adv.Mater.21 4593
[4]Jain A,Hautier G,Moore C J,Ong S P,Fischer C C,Mueller T,Ceder G 2011 Comput.Mater.Sci.50 2295
[5]Wu M S,Xu B,Ouyang C Y 2016 Chin.Phys.B 25 018206
[6]Knauth P 2009 Solid State Ionics 180 911
[7]Takada K 2013 Acta Mater.61 759
[8]Yao X,Huang J,Yin J,Peng G,Huang Z,Gao C,Liu D,Xu X 2016 Chin.Phys.B 25 018802
[9]Tatsumisago M,Nagao M,Hayashi A 2013 J.Asian Ceram.Soc.1 17
[10]Huggins R A 1999 J.Power Sources 81–82 13
[11]Chen Z H,Christensen L,Dahn J R 2003 Electrochem.Commun.5 919
[12]Xiao R J,Li H,Chen L Q 2015 J.Materiomics 1 325
[13]Anurova N A,Blatov V A 2009 Acta Crystallogr.B 65 426
[14]Brown I D 2009 Chem.Rev.109 6858
[15]Adams S,Prasada Rao R 2011 Phys.Status Solidi A 208 1746
[16]Meng Y S,Elena Arroyo-de Dompablo M 2009 Energy Environ.Sci.2 589
[17]Xiao R J,Li H,Chen L Q 2015 Sci.Rep.5 14227
[18]Wang D,Zhang X,Xiao R J,Lu X,Li Y,Xu T,Pan D,Hu Y S,Bai Y 2018 Electrochim.Acta 265 244
[19]Kamaya N 2011 Nat.Mater.10 682
[20]Mizuno F,Hayashi A,Tadanaga K,Tatsumisago M 2005 Adv.Mater.17 918
[21]Mo Y,Ong S P,Ceder G 2012 Chem.Mater.24 15
[22]Tachez M,Malugani J P,Robert G 1984 Solid State Ionics 14 181
[23]Wang X L,Xiao R J,Li H,Chen L Q 2016 Phys.Chem.Chem.Phys.18 21269
[24]Chen Y,Xi X,Yim W L,Peng F,Wang Y,Wang H,Chen Z 2013 J.Phys.Chem.C 117 25677
[25]Zhang X,Wang Y,Lü J,Zhu C,Li Q,Zhang M,Li Q,Ma Y 2013 J.Chem.Phys.138 114101
[26]Zhong X,Wang H,Zhang J,Liu H,Zhang S,Song H F,Yang G,Zhang L,Ma Y 2016 Phys.Rev.Lett.116 057002
[27]Wang Y,Lü J,Zhu L,Ma Y 2012 Comput.Phys.Commun.183 2063
[28]Wang X L,Xiao R J,Li H,Chen L Q 2017 Phys.Rev.Lett.118 195901
[29]Ward L,Agrawal A,Choudhary A,Wolverton C 2016 npj Comput.Mater.2 16028
[30]Mueller T,Kusne A G,Pamprasad R 2016 Rev.Comput.Chem.29 186
[31]Ghiringhelli L M,Vybiral J,Levchenko S V,Draxl C,Scheffler M 2015 Phys.Rev.Lett.114 105503
[32]Rupp M,Tkatchenko A,Muller K R,Anatole von Lilienfeld O 2012 Phys.Rev.Lett.108 058301
[33]Artrith N,Urban A 2016 Comput.Mater.Sci.114 135[34]Wang Y S,Yu X Q,Su S Y,Bai J M,Xiao R J,Hu Y S 2013 Nat.Commun.4 2365
[35]Wang X L,Xiao R J,Li H,Chen L Q 2017 J.Materiomics 3 178
