拓撲半金屬材料的單晶生長研究進展?
2018-07-10 09:32:42伊長江1王樂1馮子力1楊萌1閆大禹1王翠香1石友國1
物理學報 2018年12期
關鍵詞:生長
伊長江1)2) 王樂1)2) 馮子力1)2) 楊萌1)2) 閆大禹1)2)王翠香1)2) 石友國1)2)?
1)(中國科學院物理研究所,北京凝聚態物理國家研究中心,北京 100190)
2)(中國科學院大學物理科學學院,北京 100049)
(2018年4月24日收到;2018年5月8日收到修改稿)
1 引 言
一般地,按照能帶交叉點的簡并情況,可以將拓撲半金屬材料分為拓撲Dirac半金屬、Weyl半金屬和Node-Line半金屬等[1?3]. 目前,已經有許多理論預測的拓撲半金屬材料被成功地制備出來并得到驗證. 比如,Dirac半 金 屬Na3Bi[4?6],Cd3As2[7?9]等;Weyl半 金屬TaAs family[10?12],Mo/WTe2[13?17]等;Node-Line半 金 屬ZrSiS[18?20],PbTaSe2[21?23],TiB2family[24?26]等.此外,近年來還出現了新型的拓撲材料,如含有“沙漏型”費米子的拓撲晶體絕緣體材料KHgSb[27?29],含有三重簡并點的拓撲半金屬MoP,WC[30?33]等.
對于拓撲半金屬材料,理論研究一直處于領先的位置,并且已經形成了由理論預測到材料單晶制備再到角分辨光電子能譜儀(ARPES)驗證以及電磁輸運性質測量佐證的研究模式.由于ARPES實驗驗證材料拓撲性的對象必須是單晶材料(包括單晶塊材以及單晶薄膜),因此,在整個研究過程中,材料的單晶生長是必不可少的一個環節.另外,拓撲半金屬材料也擁有奇特的電磁輸運性質,比如量子自旋霍爾效應、量子振蕩以及由手性異常導致的負磁阻現象等,單晶材料在這些研究中同樣也有著不可替代的地位.
一般地,制備單晶的方法主要有助熔劑法、氣相傳輸法、水熱法、提拉法以及高壓合成等方法.針對材料所具備的化學性質,需要采用不同的方法來進行合成.常用的方法主要有兩種:助熔劑法和化學氣相傳輸法.本文首先介紹主要用到的兩種單晶生長方法的原理和過程,然后再詳細介紹幾種拓撲半金屬單晶的生長方法.
2 單晶生長方法
2.1 助熔劑法
傳統生長晶體的方法主要為高溫熔融冷凝結晶,這種方法需要的溫度一般很高,而且需要特殊的耐熱材料作為載體,因而導致成本和能耗都比較高.助熔劑法是一種能夠通過低熔點物質來溶解高熔點物質,從而降低結晶溫度的一種方法.除了能夠降低反應物的熔點之外,由于晶體在溶液中自由生長而沒有大的溫度梯度,所以它們通常顯示出自然生長面,可以生長出良好成形和高質量的晶體.另外,這樣生長的單晶通常缺陷比較少,均一性好.但是,生長過程中助熔劑以及坩堝材料等可能會進入到晶體內部造成污染,因此,選擇合適的助熔劑和合適的坩堝材質,能夠有效解決這個問題[34,35].
助熔劑的種類很多,金屬單質、合金、低熔點氧化物以及鹽等,都可以作為助熔劑[36?38].常見的金屬單質助熔劑有Al,Ga,In,Sn,Pb,Bi和Zn等低熔點金屬.另外像一些熔點較高的Fe,Co,Ni和Cu等金屬也可以作為助熔劑來溶解熔點更高的物質,如Ta,W和C等.金屬單質助熔劑是最常用的助熔劑.金屬合金助熔劑主要有CuAs合金、CuP合金等.氧化物助熔劑比較多,常用的有Bi2O3,B2O3,PbO等.鹽類助熔劑主要有NaCl,KCl,RbCl等.通過相圖指導以及大量的實驗摸索,可以找到晶體生長所需的最佳助熔劑.除了助熔劑之外,對于坩堝的選擇也要視反應物化學性質而定.金屬單質、合金或鹽助熔劑一般采用Al2O3坩堝;如果需要堿金屬或堿土金屬作助熔劑時,可以采用Mo坩堝;氧化物助熔劑一般采用Pt坩堝.圖1(a)所示為常用的氧化鋁坩堝、Mo坩堝、Pt坩堝、Ta管以及石英管等.
助熔劑法除了選擇最佳的助熔劑和化學性質穩定的坩堝外,對于溫度控制的要求也很高.現在商業化的電阻爐已經能夠精確控制溫度,提供優良的保溫以及降溫條件.由于金屬或者合金助熔劑在高溫時容易氧化,因此需要進行封管操作來隔絕氧氣.一般有石英管和鉭管兩種封管選擇,視不同的情況而定.石英管保護的最高溫度為1200?C左右,當需要的反應溫度更高時,則要采用氣氛保護的方法.如圖1(b)—(e)所示,依次為井式爐、單溫區管式爐、雙溫區管式爐及氣氛保護箱式爐.

圖1 用于生長單晶的各種坩堝、石英管以及電爐 (a)由左至右依次為Ta管、Pt坩堝、氧化鋁坩堝、Mo坩堝以及石英柱和石英管;(b)—(e)分別為井式爐、單溫區管式爐、雙溫區管式爐及氣氛保護爐Fig.1.Crucibles,quartz tubes and furnaces.(a)From left to right:Ta tubes,Pt crucible,alumina crucibles,Mo crucibles,silica plugs and silica tubes.(b)–(e)Pit furnace,single-temperature zone tube furnace,double temperature zone tube furnace,and atmosphere furnace.
2.2 氣相傳輸法
氣相傳輸法包括物理氣相傳輸和化學氣相傳輸法.兩種過程均需要一定的溫度梯度,一般在水平放置的管式爐中進行.
物理氣相傳輸法是指一些具有高蒸氣壓的材料,在到達一定溫度時會升華成為氣態,如果將材料密封在管子中(通常用石英管),并放置在具有適當溫度梯度的水平管式爐中,則晶體會沉積在密封管中的不同位置處.
化學氣相傳輸法則要借助一些揮發性材料作為反應載體(常見載體材料有I2,AlCl3,TeCl4等),將原材料從一端傳輸至另一端結晶,這中間傳輸載體會和原材料發生化學反應,然后在溫度的驅動下再脫離原材料,返回原始成分.原材料通常位于熱區,晶體沉積在冷區,但也有可能相反,這取決于反應的熱力學和動力學[39?41].
3 Dirac半金屬的單晶生長
自三維Dirac材料被理論預言以來就獲得了非常廣泛的關注.Na3Bi,Cd3As2以及EuCd2As2等材料的提出,打開了在固體材料中探尋三維Dirac費米子的大門.Na3Bi和Cd3As2是首先被預言的三維Dirac材料[4,7],其中的Dirac點剛好處在費米能級附近,是非常理想的三維Dirac材料.EuCd2As2被認為是同時打破了空間及時間反演對稱性卻受到空間-時間聯合對稱性所保護的新型Dirac材料[42].這幾種材料的多晶形態已經被研究,結構也早已被人們認知.理論上只要存在這種材料,就能夠生長出其單晶形態.上述的材料均能夠用助熔劑法長出高質量、大尺寸的單晶.
3.1 Na3Bi
Na3Bi是六方層狀的化合物,幾十年前就已經被發現并確定了結構[43],其晶胞參數為a=5.448 ?,c=9.655 ?. 通過觀察材料的化學式,發現Na和Bi都是熔點很低的金屬,分別為98?C和271?C,但是Na3Bi的熔點反而很高,為845?C.同時,在Na比例比較多時,并沒有其他化合物產生,如圖2(c)所示[44].因此,Na是生長這個單晶的理想助熔劑.Kushwaha等[45]用兩種方法分別生長了Na3Bi的單晶材料,直接熔融結晶法長出的單晶如圖2(a)所示,沒有非常好的解離面,而是形成了金屬錠子一樣的單晶,限制了后續的測試實驗.利用Na作為助熔劑長出的單晶如圖2(b)所示,呈現出非常平整的六角結構的晶體表面,這也是Na3Bi的自然解離面.我們也采用Na助熔劑方法生長了Na3Bi的單晶,得到了六角形片狀的單晶材料,如圖2(d)所示.對于這個材料的生長,有以下4點需要注意:1)金屬Na極易氧化,因此要在極低氧、極低水含量并且有氬氣保護的手套箱中進行操作;2)金屬Na高溫下會揮發,產生的Na蒸氣會腐蝕石英管,因此要封入Ta管中;3)Na3Bi單晶的結構中含有單層的Na原子,從而導致其對氧氣十分敏感,因此晶體需要在手套箱中進行處理;4)離心后大量的Na需要妥善處理,防止意外發生.

圖2 Na3Bi單晶照片及Na-Bi二元相圖 (a),(b)Kushwaha等生長的Na3Bi單晶[45];(c)Na-Bi的二元相圖[44];(d)本文生長的Na3Bi單晶Fig.2.Photograph of Na3Bi single crystals and binary phase diagram of Na-Bi system:(a),(b)Single crystals of Na3Bi grown by Kushwaha et al.[45];(c)binary phase diagram of Na-Bi system[44];(d)single crystals of Na3Bi grown by this work.
3.2 Cd3As2
同Na3Bi一樣,Cd3As2也是理論預言的三維Dirac半金屬材料,雖然是二元化合物,但由于Cd3As2材料擁有多個不同的結構相,因此在生長單晶時,需要對助熔劑的用量和反應溫度進行精確控制.理論預言空間群為I41cd的結構相,也就是其低溫相,才擁有Dirac費米子[7].通過相圖可知[46],當溫度低于578?C時,才會產生低溫相(如圖3所示).采用Cd作助溶劑,Cd和As的摩爾比約為9:1.放入9倍摩爾比的Cd的原因是可以在降溫時避免析出高溫相的晶體,并直接析出低溫相的晶體.然后將混合物裝入氧化鋁坩堝并封入石英管,升溫至900?C保持10 h,然后緩慢降溫至500?C離心,可以得到大尺寸的、具有光潔的生長面的單晶體(如圖3中的插圖所示).

圖3 As-Cd二元相圖,其中插圖所示為Cd3As2的單晶照片以及局部放大圖[46]Fig.3.Binary phase diagram of As-Cd system.The inset is a photograph and its detail view of Cd3As2 single crystals[46].
生長Cd3As2單晶時需要注意的兩個問題:1)As元素具有較大的蒸氣壓,升溫速率應當比較慢;2)Cd元素和As元素都是對人體有害的元素,要妥善處理原材料以及離心后多余的助熔劑廢料.
3.3 EuCd2As2
固體中的Dirac費米子是受到空間反演對稱性和時間反應對稱性保護的,一旦打破其中一種對稱性保護,就會形成Weyl費米子.而EuCd2As2被認為是打破了空間反演對稱(inversion symmetry P)和時間反演對稱性(time-reversal symmetry T),但沒有打破空間-時間聯合對稱性(combined PT symmetry)的Dirac半金屬材料[42].EuCd2As2是六方層狀結構的材料,空間群是P-3m1,其中Eu形成三角格子層.
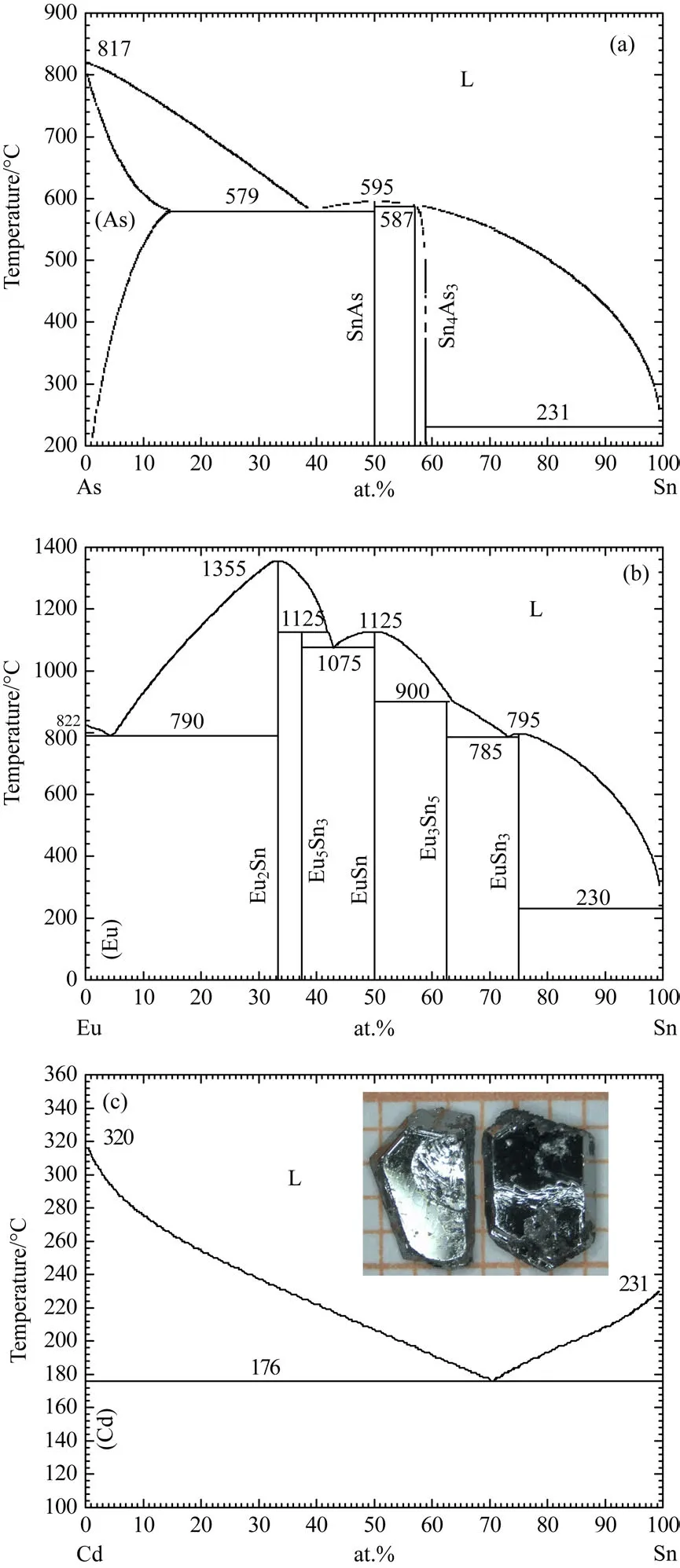
圖4 (a)—(c)分別為As-Sn,Eu-Sn和Cd-Sn的二元相圖;(c)中的插圖為EuCd2As2的單晶照片[47?49]Fig.4.(a)–(c)Binary phase diagrams of As-Sn,Eu-Sn and Cd-Sn systems,respectively.The insert is a photograph of EuCd2As2single crystals[47?49].
首先,在這個材料中,金屬Cd的熔點最低,只有321?C,所以直接用Cd來做助熔劑進行單晶生長是最佳選擇.我們進行了多次嘗試,通過調整元素的摩爾比例以及反應溫度來摸索最優的生長條件.但是我們發現,利用Cd作助熔劑雖然能夠生長出EuCd2As2單晶,但是長出的單晶尺寸都很小,約0.1 mm大小,很難用于電磁輸運測量實驗和ARPES實驗.經過調研,我們發現Sn是比較好的助熔劑,圖4(a)—(c)所示分別為As-Sn,Eu-Sn和Cd-Sn的二元相圖[47?49].首先Sn的熔點低,容易進行離心;其次,通過控制摩爾比例,可以避免Sn和這三種元素形成化合物,如Eu3Sn5,EuSn3等雜相.根據相圖以及實驗條件摸索,最終確定Eu:Cd:As:Sn=1:2:2:10的摩爾比例.溫度控制程序為:10 h升溫到1000?C并維持20 h,然后以2?C/h的速率降溫至500?C,最后離心得到單晶.這樣生長出的晶體尺寸可達到1 mm×3 mm×4 mm,并且擁有非常光潔的六角形表面,如圖4(c)中的插圖所示.但是這種方法有個非常大的缺點就是有可能會引入Sn雜質.
3.4 Ir1?xPtxTe2(0 6 x 6 0.5)
IrTe2材料是六方層狀的,早先的研究表明該材料中存在一個結構相變,而且在摻雜Pt之后會壓制相變并在低溫誘導出超導性[50].在類似結構的材料中,六方PdTe2[51,52],PtSe2[53,54]以及PtTe2[55]等材料中均有報道稱存在第二類Dirac點.其中PdTe2還具有超導電性,但是由于PdTe2中的Dirac點距離費米面比較遠,因此可認為其中的超導電性與Dirac電子之間的關系尚未定論.IrTe2材料擁有類似的晶體結構和電子結構,理論計算發現其中也存在第二類Dirac電子,并且通過摻雜多一個外電子的Pt元素,能夠保證結構不變的情況下連續地調控Dirac點的位置.摻雜Pt不僅將Dirac點調控至費米面附近,同時還誘導出了超導電性,因此Ir1?xPtxTe2被認為其超導電性可能會與Dirac電子有關[56,57].
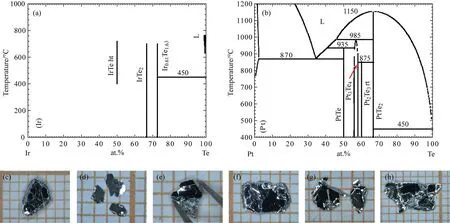
圖5 (a),(b)分別為Ir-Te和Pt-Te的二元相圖[58,59];(c)—(h)為Pt的名義摻雜濃度x=0.05,0.1,0.25,0.3,0.4,0.5時的Ir1?xPtxTe2單晶樣品Fig.5.(a)–(b)Binary phase diagram of Ir-Te and Pt-Te systems[58,59];(c)–(h)single crystals of Ir1?xPtxTe2for nominal doping concentration of x=0.05,0.1,0.25,0.3,0.4,0.5.
Ir和Pt都是熔點特別高的元素,如圖5(a)和圖5(b)相圖所示,Te元素能夠溶解這兩種金屬[58,59].Te元素的熔點較低,而且這個材料本身含有Te,因此Te對于這個材料來說是一種非常好的助熔劑.但是,需要注意的問題是,Ir-Te的化合物中存在Ir3Te8這種材料.為避免長出Ir3Te8,需要對物質的摩爾比以及反應溫度和離心溫度進行合理的控制.經過探索發現,當摩爾比為Ir1?xPtx:Te=1:8時,燒結溫度到1100?C,然后緩慢降溫至800?C離心,會得到擁有六方層狀外形的Ir1?xPtxTe2單晶.圖5(c)—(h)所示依次為名義摻雜量x=0.05,0.1,0.25,0.3,0.4,0.5時的單晶照片.這種生長方法不僅能夠得到Ir1?xPtxTe2單晶,也會伴隨著產生Ir3Te8的單晶體,不過可以在光學顯微鏡下根據晶體外形做出判斷.在實驗過程中,為保證單晶的質量以及摻雜的均一性,我們將Ir和Pt按照一定的比例進行電弧熔煉,除去粉末中的其他雜質,將得到的合金剪成碎塊,然后再和Te塊混合進行燒結.通過這種方法可以實現對摻雜量的精準調控,進而對Dirac點的位置進行精確調控.
4 Weyl半金屬的單晶生長
固體材料中的Dirac費米子在空間反演對稱性和時間反演對稱性的保護下是四重簡并的,當打破其中一種對稱性時,Dirac點會演化為一對兩重簡并且手性相反的Weyl點.理論預言TaAs,TaP,NbAs和NdP[10?12]材料中存在打破空間反演對稱性的Weyl點,并由實驗證實.另外還有理論預言在正交結構的Mo/WTe2[13?17]中存在第二類Weyl費米子.TaAs家族的單晶生長已有詳細的報道[60,61],通過化學氣相傳輸的方法,可以生長出較大尺寸的單晶材料,本文不再做進一步討論.HgCr2Se4,Y2Ir2O7和ZrCo2Sn系列材料是被理論預言的另外一類打破時間反演對稱性的磁性Weyl費米子材料[62?64],這類材料中一般都存在順磁-鐵磁相變,當材料處于鐵磁相中時,會演化出Weyl點.HgCr2Se4和ZrCo2Sn系列的單晶材料已經可以生長出來,但是Y2Ir2O7的單晶材料還未成功生長出.
4.1 Mo/WTe2
正交結構的MoTe2材料的結晶溫度點比較高,并且當溫度低于820?C左右時,就會發生結構相變,轉變為低溫六方相.由圖6(a)所示的相圖可知[65],Te作助熔劑時,需要很高的摩爾比例才能使得Mo溶解在Te中.針對金屬Mo的特點,我們采用Te助熔劑法和化學氣相傳輸兩種方法進行嘗試.首先是助熔劑法,將高純的Mo粉末和Te塊材按照摩爾為1:20的配比裝入氧化鋁坩堝并封入石英管中.溫度控制程序為10 h升溫到1100?C并維持10 h,然后以2?C/h的速率降溫至950?C,并在這個溫度進行離心.經過幾次實驗條件探索,用助熔劑法能夠生長出尺寸最大能達到0.2 mm×2 mm×6 mm的矩形片狀單晶,并且單晶擁有非常明亮的金屬反光,這說明晶體擁有比較平整的自然生長面,圖6(b)所示為相應的晶體照片.另外,我們還利用化學氣相傳輸法進行了嘗試,利用I2單質作為傳輸介質,高溫端設置為1000?C,溫度梯度約為100?C,利用這種方法生長出的晶體尺寸比較小,而且沒有光潔的表面.
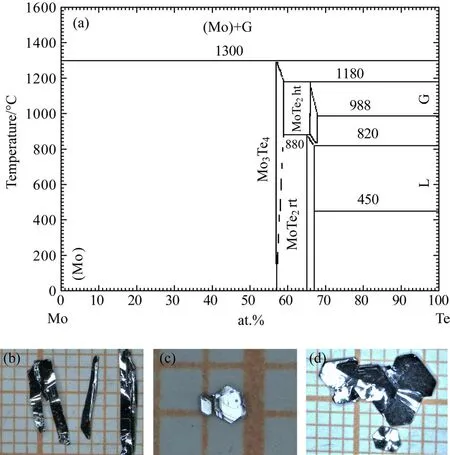
圖6 (a)Mo-Te二元相圖[65];(b)助熔劑法生長的正交相MoTe2;(c)助熔劑法生長的六方相MoTe2;(d)氣相傳輸法生長的六方相MoTe2的單晶照片Fig.6.(a)Binary phase diagram of Mo-Te system[65];(b)single crystals of orthorhombic-MoTe2grown by fl ux method;(c)single crystals of hexagonal-MoTe2 grown by fl ux method;(d)single crystals of hexagonal-MoTe2grown by vapor transport method.
除了生長正交結構的MoTe2單晶之外,我們也嘗試生長了低溫六方相的MoTe2.嘗試用兩種方式進行低溫相MoTe2單晶的生長,利用Te作助熔劑和利用I2單質作為傳輸介質做氣相傳輸.利用Te作助溶劑時,和上述配比一樣,只不過慢降溫至600?C離心,這樣能夠得到小塊的六方形狀的MoTe2晶體,如圖6(c)所示.然后嘗試用氣相傳輸的方法,將總質量為3 g的Mo粉和Te粉按照摩爾比1:2充分研磨,在500?C保溫48 h作為前驅體.將前驅體在手套箱中再次研磨后,加入0.3 g的I2作為傳輸介質,封入石英管內.石英管的長度為15 cm,內徑約為2 cm.將石英管放入雙溫區管式爐,熱端溫度為800?C,冷端為750?C,維持20 d.通過這種方法長出的單晶有明顯的六方片狀外形,如圖6(d)所示,尺寸最大可達到0.1 mm×4 mm×4 mm.
WTe2成為熱點材料是因為在其中發現了巨大的磁電阻效應[66].該材料的單晶生長和MoTe2的過程大概相同.W金屬同樣擁有比較高的熔點,并且在Te中的溶解度非常低[67].不過當Te的比例很高時,不會產生別的化合物,反應產物會比較干凈,如圖7所示.同樣采用Te助熔劑法來生長這個材料.將W粉末和Te塊材按照W:Te=1:30的摩爾比例封入氧化鋁坩堝并封入石英管中,加熱至1100?C維持10 h,并緩慢降溫到800?C后離心,能夠得到尺寸約為0.2 mm×1 mm×5 mm的長條層狀單晶材料,圖7中的插圖為采用該方法生長出的WTe2單晶.這種方法能夠生長出Te缺陷比較少的WTe2單晶,而且晶體表面十分平整.但是這種方法的晶體產量比較低,需要消耗大量的原材料.
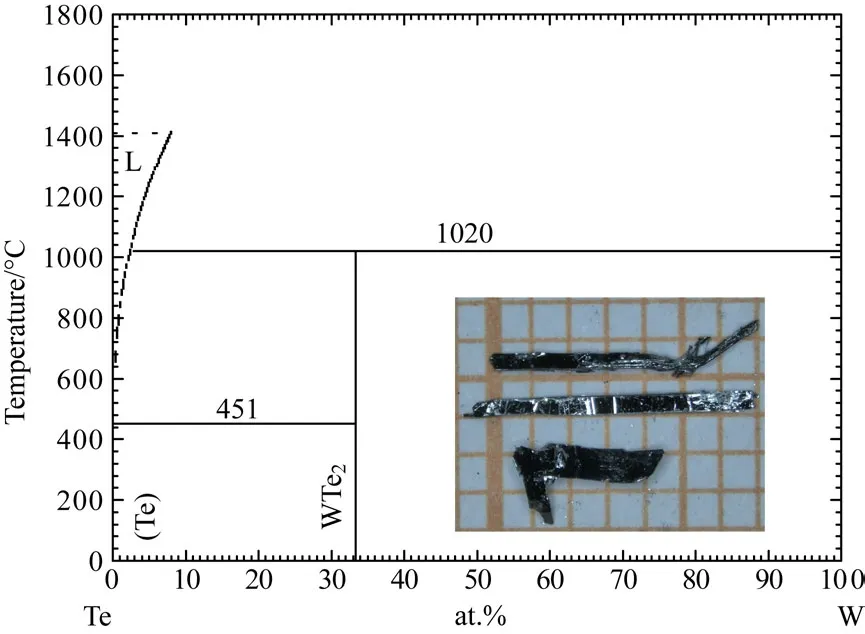
圖7 Te-W二元相圖,插圖所示為WTe2的單晶照片[67]Fig.7.Binary phase diagram of Te-W system.The inset is a photograph of WTe2single crystals[67].
4.2 HgCr2Se4
HgCr2Se4和Y2Ir2O7材料是最早被預言的磁性Weyl費米子材料[62,63].理論計算指出在HgCr2Se4中只存在一對Weyl點,且位于費米能級附近. 以前的研究發現HgCr2Se4是一個鐵磁半導體,最近對于其單晶輸運性質的測量發現,HgCr2Se4在其鐵磁態擁有近97%的自旋極化率[68],并且在順磁-鐵磁相變點處表現出極大的負磁阻現象[69].遺憾的是,在輸運性質以及ARPES測試中尚未發現Weyl點存在的證據.
觀察HgCr2Se4的化學式會發現,其中Hg和Se都有揮發性以及比較高的蒸氣壓,Cr具有較高的熔點,因此采用助熔劑法有兩個明顯的缺點:1)助熔劑法需要比較高的溫度環境,高溫下Hg和Se的蒸氣壓高,容易爆管;2)引入第四種元素作助熔劑,會引入其他雜質化合物.HgCr2Se4的單晶早在1969年就已經通過化學氣相傳輸法生長出來[70,71],利用AlCl3和CrCl3作傳輸介質,能夠生長出其單晶材料.首先,將Hg,Cr和Se按照1:2:4的摩爾比進行配比,總質量約為3 g,在300?C維持30 h作為前驅體材料.然后加入約1 g的AlCl3或者CrCl3粉末,混合后再次封入到石英管中.將抽真空并且密封的石英管放入水平單溫區管式爐中,熱端為原材料,溫度設置為800?C,溫度梯度約為100?C.經過10 d的反應時間,能夠得到尺寸為1.5 mm×1.5 mm×1.5 mm左右,具有八面體外形以及光潔表面的單晶體,如圖8(a)所示.

圖8 (a)HgCr2Se4和(b)ZrSiS的單晶照片Fig.8.Photographs of(a)HgCr2Se4and(b)ZrSiS single crystals.
5 Node-Line半金屬的單晶生長
Node-Line半金屬材料是另外一種具有奇特電子態的拓撲材料.在動量空間中,如果能帶節點組成一個環或者是周期性連續的線,這種材料就被稱為Node-Line材料.理論預言的Node-Line材料有Cu3PdN[72],ZrSiS[18]和TiB2家族[24]等材料.迄今為止,對于反鈣鈦礦結構的Cu3PdN材料,始終沒有關于其單晶塊材的報道,但是對其單晶薄膜的生長和研究已有報道[73].本部分我們將著重介紹ZrSiS和TiB2這兩個材料的單晶生長.
5.1 ZrSiS
理論預言ZrSiS家族(Zr可以替換為Hf;Si可以替換為Ge,Sn;S可以替換為Se,Te)是一類Dirac型Node-Line半金屬材料.ARPES實驗在ZrSiS中發現了Node-Line形式的電子態[20,74].同時電磁輸運性質的測量表明[19,75],ZrSiS單晶擁有非常大的磁電阻,并且在低溫高磁場的條件下還會出現量子振蕩,這也間接證明了其中Node-Line費米子態的存在.
ZrSiS材料可以通過物理氣相傳輸的方法進行生長.首先將Zr,Si和S的粉末按照摩爾比為1:1:1配比,稱量總質量約為3.5 g.充分研磨混合后,封入石英管中燒結制備多晶.在此需要注意,單質S在高溫下的蒸氣壓很大,有爆管的危險性.因此我們采取了兩段式升溫:先以100?C/h的速率升溫到400?C(S單質的沸點是444?C),然后維持20 h;再以50?C/h的速率升溫至1000?C保持100 h.將燒結完成的ZrSiS多晶研磨成粉,然后加入0.3 g的I2單質作為傳輸介質,封入石英管中.石英管的長度為12 cm,內徑為1.7 cm,在裝入反應原料之前,石英管在抽真空的狀態下用煤氣氧氣火焰噴槍烘烤,清除表面吸附的雜質.將石英管封裝好之后,置于水平放置的單溫區管式爐中,熱端溫度設置為1050?C,溫度梯度約為100?C,反應時間為10 d.在石英管的冷端長出了尺寸約為2 mm×2 mm×0.2 mm的四方形狀的單晶,如圖8(b)所示.在收集單晶時我們發現,未完全被傳輸到冷端的多晶粉末其實也已經結晶成了很小的單晶體.將這些細碎的晶體在手套箱中繼續研磨成粉末,然后重復氣相傳輸,發現依然能夠得到尺寸較大并且結晶性很好的單晶體.這一點可以說明ZrSiS的結晶過程是一個物理過程,傳輸介質I2作為載體,將ZrSiS分子搬運至冷端結晶.
5.2 TiB2
TiB2系列材料包括ZrB2和HfB2,都是AlB2形式的六方結構的材料.最近,理論預言在不考慮自旋軌道耦合的情況下,這一類材料中含有Dirac型的Node-Net電子態,它的費米面是由四種Node-Line相連接組合成的網狀結構[24].Ti和B都是熔點非常高的元素(Ti的熔點為1668?C,B的熔點為2076?C),因此生長這個單晶具有一定的挑戰性.早期已有對這一系列材料單晶生長的報道,利用Co,Fe或者Al作為助熔劑都能夠生長出其單晶形態[76,77].最近有報道稱在TiB2中觀測到Node-Net電子態[25]以及在ZrB2中觀測到巨大的磁電阻現象[26].下面主要論述利用Al作助溶劑生長TiB2單晶的方法.
圖9(a)所示為Al-B的二元相圖[78],可以看到當Al的摩爾數約為B元素的10倍時,溫度燒至1300?C即可讓B元素溶解在Al的高溫液體中.并且此時Al的摩爾量為Ti的20倍,根據圖9(b)所示的Ti-Al相圖[79],可以得到這個倍數的Al也能夠溶解Ti,因此采用Al作助熔劑.我們將Ti柱、B顆粒和Al顆粒按照1:2:20的比例分別混合后,放入氧化鋁坩堝中,并在氬氣氛圍保護的硅鉬棒電爐中進行燒制.反應溫度程序為:15 h加熱至1500?C并維持20 h,然后以1?C/h的速率降溫至1300?C后將電爐關閉,自然降溫到室溫.待降到室溫后,將裝有產物的氧化鋁坩堝取出并封入到石英管中,重新加熱至900?C離心,將多余的Al助熔劑去除.這里由于助熔劑的量太大,所以不采用鹽酸或者熱濃堿溶液直接浸泡的方法,而是先離心出大部分的助熔劑,然后再用熱的濃堿溶液進行浸泡,去除晶體表面的Al助熔劑.通過這種方法能夠得到尺寸約為0.2—0.4 mm大小的TiB2單晶材料,同時也有大量的AlB2單晶(六方片狀)、少量的TiAl3單晶 (四方片狀)和AlBx(顆粒狀棕色透明晶體)單晶出現.雖然通過觀察外形可以在光學顯微鏡下挑選出TiB2單晶,但是這種方法TiB2單晶的產量特別低,Ti和B的利用率也較低.為解決這個問題,我們增加了B的比例,將Ti,B和Al按照1:4:40和1:6:60的比例混合后,按照原始溫度控制程序進行燒制.結果在Ti:B=1:6的配比下,AlB2和TiAl3的晶體含量明顯減少,產物中六方片狀的TiB2和顆粒狀棕色透明的AlBx晶體的數量增加,并且在光學顯微鏡下容易區分.通過這種原材料配比得到的TiB2單晶的尺寸也比較大,達到0.8—1 mm,而且有些單晶能夠保持完整的六方形狀,如圖9(a)中的插圖所示.
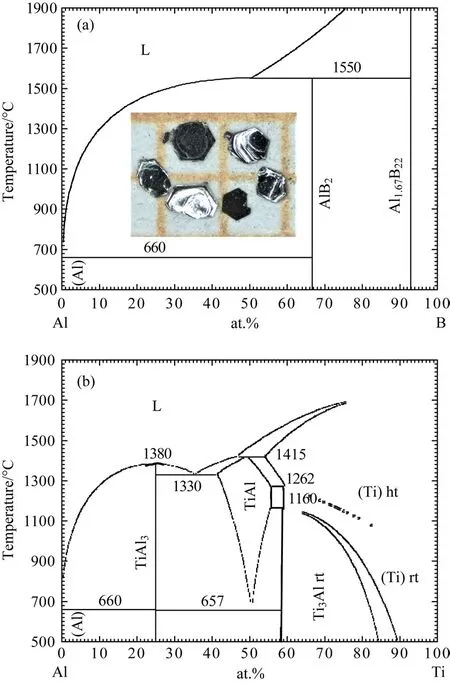
圖9 (a),(b)分別為Al-B和Al-Ti的二元相圖,(a)中的插圖為TiB2的單晶照片 [78,79]Fig.9.(a),(b)Binary phase diagram of Al-B and Al-Ti systems;the inset of(a)is photograph of TiB2 single crystals[78,79].
6 其他拓撲材料
對于拓撲半金屬材料按照其中費米子的簡并態可以分類分為Dirac半金屬、Weyl半金屬和Node-Line半金屬.但是最近的理論研究發現,除了這三種費米子之外,還有其他的新型的拓撲態下的費米子,比如在KHgSb中發現的“沙漏型費米子”[27];在MoP,WC系列材料中發現的三重簡并的新型費米子等[30].另外,還有一些預言的其他新費米子材料,比如八重簡并的Ba4Bi3和La4Bi3以及三重簡并的Pd3Bi2S2等[80].我們首次生長出了KHgSb和MoP的單晶材料,并與理論計算課題組和ARPES實驗課題組進行合作,首次在實驗上證明了這兩種新奇費米子的存在.下面將詳細介紹這兩種材料的生長方法.
6.1 KHgSb
多晶材料的KHgSb早在1980年就被合成出來[81],由于這個材料對空氣極其敏感,因此沒有太多關于其物理性質的報道.對這個材料理論計算的報道有兩種觀點:一是認為KHgSb是弱拓撲絕緣體[28];二是認為其中含有“沙漏費米子”[27].我們成功生長出了這個材料的單晶,并對其結構和拓撲性進行了系統表征,結合ARPES實驗,找到了其中存在“沙漏費米子”的證據[29].
通過調研相圖[82?84],我們決定采用自助熔劑的方法來生長KHgSb.生長KHgSb的難點在于對金屬K和Hg的處理,金屬K的熔點比較低,常溫下為黏滯性比較大的固體,并且高溫蒸氣壓也比較大,更重要的一點是它會和成分為SiO2的石英管發生反應;金屬Hg的蒸氣壓更大,而且Hg常溫下為液態,在稱量時不容易控制,并且Hg是有毒的材料,需要妥善處理.為了保護石英管不被K腐蝕以及因為蒸氣壓過高而炸裂,我們采用了容積約18 mL的Ta管作為保護.首先,將總質量為4 g左右的K,Hg和Sb按照2:1:1的摩爾比例進行配置,其中多余的K起到助熔劑的作用.在配置過程中,K和Hg元素會立即互溶在一起,并釋放出大量熱量,因此操作時應避免燙傷.將裝有原材料氧化鋁坩堝利用電弧焊密封在Ta管中,然后再將焊好的Ta管封入石英管中,此時的石英管的作用是為了避免高溫下Ta管的氧化.將封好的石英管放入馬弗爐內,5 h加熱至200?C并維持20 h,然后10 h升溫至800?C并維持10 h,之后以2?C/h的速率降溫至400?C,并在400?C保溫退火10 d后,在冰水中淬火.燒制完成后將Ta管取出,放入到手套箱中,在手套箱中解開.通過這種方法可以得到表面平整光亮的、并且有六方形狀的片狀單晶.很遺憾,由于測試時間倉促,我們沒有留下晶體的照片記錄.
我們對得到的單晶進行了結構表征,證實了得到的單晶正是我們想要的材料.圖10(a)和圖10(b)是根據KHgSb的結構解析得到的結構示意圖,能夠看出明顯的層狀結構.圖10(c)是KHgSb單晶沿著(001)方向的X射線衍射譜,可以看出得到的單晶體擁有很好的結晶性.圖10(d)是將單晶在煤油(在煤油中研磨是為了隔絕氧氣和水分)中研磨成粉末后測到的X射線衍射圖譜,藍色是研磨后馬上快速測量的結果,可以看到除了KHgSb的主相之外,還有Sb的相.圖10(d)中的星標是Sb的衍射峰;紅色譜線是同一批粉末在15 min后測試的X射線圖,可以發現,KHgSb的相越來越弱,Sb的相越來越明顯.說明這個材料在研磨成粉末之后會慢慢分解,產生Sb單質.
ARPES實驗證實了在KHgSb單晶中存在“沙漏費米子”,KHgSb是從理論預測到單晶制備再到實驗測量驗證理論的一個典范[27,29].
6.2MoP
按照能帶簡并度來看,Dirac費米子的能帶是四重簡并的,Weyl費米子的能帶是兩重簡并的,而MoP,WC和TaN等材料[30]被預言其中存在一種全新的三重簡并的費米子.這種費米子態的存在,打破了常規的拓撲半金屬分類,也拓寬了對于拓撲材料領域的認知.
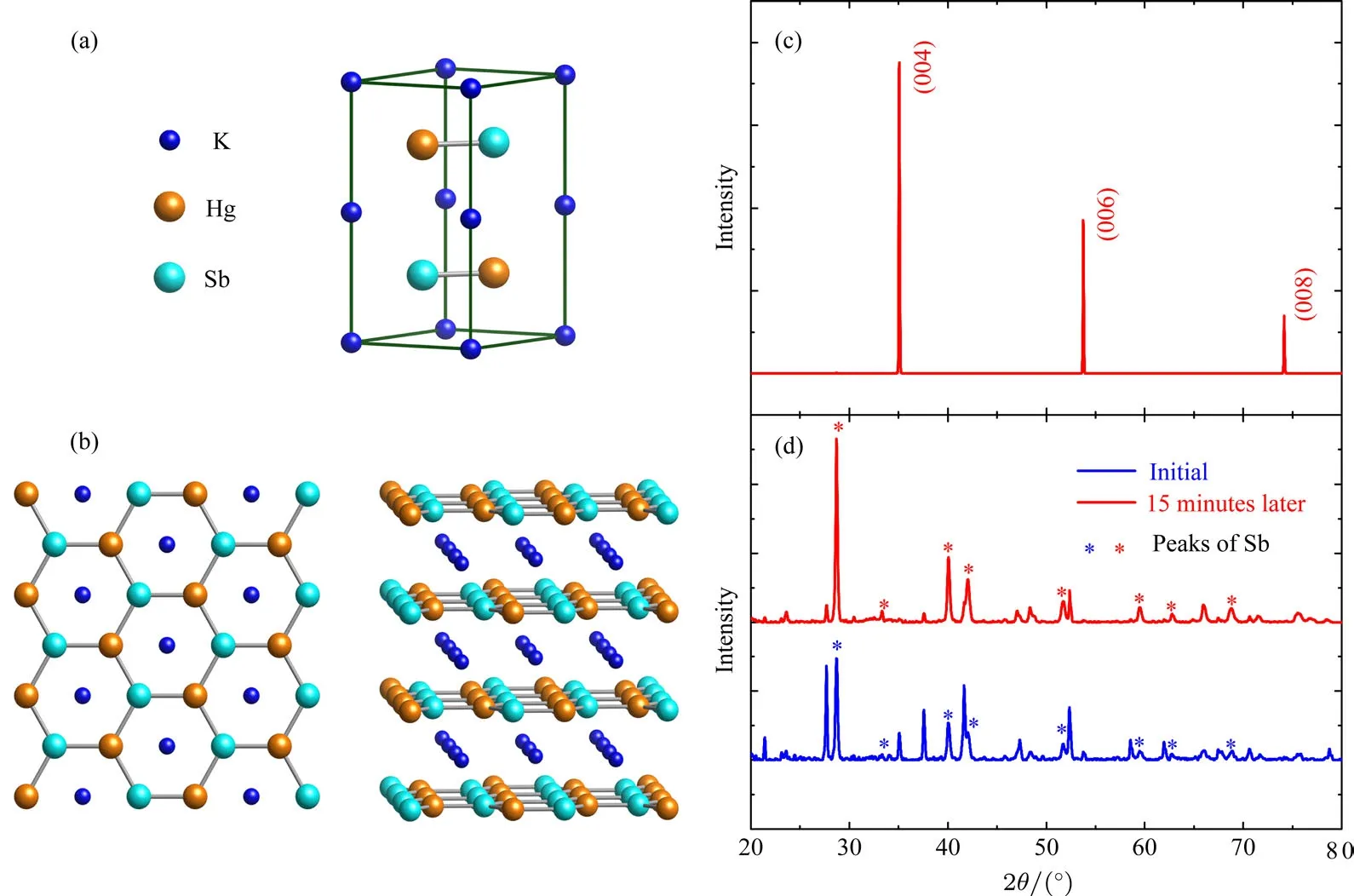
圖10 (a)KHgSb單晶的晶胞示意圖;(b)晶體沿著(001)方向的俯視圖以及三維模型圖;(c)沿著(001)方向上的晶體單面的X射線衍射圖;(d)KHgSb單晶研磨成粉末的X射線衍射圖,其中藍線代表在煤油中研磨后立即測試得到的X射線衍射圖,紅線代表經過15 min后再次測試的X射線衍射圖Fig.10.(a)The unit cell of single crystalline KHgSb;(b)a top view along c-axis and the three-dimensional model of layered structure of the crystal;(c)the X-ray di ff raction pattern of(001)surface;(d)the X-ray di ff raction pattern of KHgSb powders;the blue line represent that the measurement is immediately performed after grinding in oil;the red line represent the same progress on the same samples after 15 minutes.
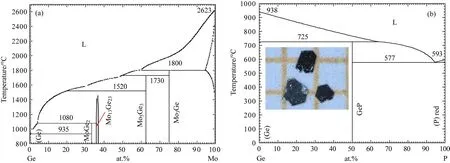
圖11 (a),(b)分別為Ge-Mo和Ge-P的二元相圖;圖(b)的內插圖為MoP的單晶照片[87,88]Fig.11.(a),(b)Binary phase diagram of Ge-Mo and Ge-P systems;the inset of(b)is a photograph of MoP single crystals[87,88].
MoP的多晶材料在幾十年前就已被合成[85],但是由于Mo的熔點很高,而且沒有合適的助熔劑來溶解Mo金屬單質.P由于在高溫下的蒸氣壓比較大等原因,一直沒有被合成單晶.Shekhar等[86]利用氣相傳輸的方法生長出了MoP的單晶,實驗方法為將Mo粉末和P粉末按照1:1配比燒制前驅體,再加入I2傳輸介質,在高溫區為1000?C,低溫區為900?C的雙溫區管式爐中燒制幾個星期,能夠得到1 mm左右的單晶.并且測量到了極低的電阻率以及量子振蕩等物理性質.這種方法雖然可以生長出MoP的單晶,但是周期卻很長.經過調研相圖,如圖11(a)和圖11(b)所示,發現Ge能夠同時溶解單質Mo和P[87,88].因此我們確定用金屬Ge作助熔劑來生長這個單晶.首先將Mo塊、P塊和Ge塊按照1:1:30的摩爾比例混合后裝入氧化鋁坩堝,Mo和P元素的總量約為2 g.然后將盛有原料的氧化鋁坩堝封入Ta管中,再將Ta管封入到石英管中.在封裝石英管時,為避免高溫下石英管變軟塌陷,要充入一些氬氣平衡氣壓.燒制溫度程序為:20 h升溫至1160?C,維持5 h,然后以1?C/h的速率緩慢降溫到1000?C,在1000?C時,將Ge助熔劑離心出來與單晶分離.通過這種方法能夠生長出最大約0.1 mm×2 mm×2 mm的六方片狀的MoP單晶材料,圖11(b)中的插圖所示為其單晶照片.我們與ARPES測試組進行合作,首次在MoP單晶中觀測到了三重簡并的費米子.此外,在高壓的條件下,能夠誘導MoP產生超導電性[89].三重簡并費米子家族的另外一個重要成員WC,可以用使用Co作助熔劑進行生長,單晶生長的過程以及物性的測量已有詳細的報道[33].
MoP中三重簡并費米子的發現打破了對拓撲半金屬材料的常規分類,對進一步研究固體中的奇特費米子具有重大的意義.同樣,MoP也是從理論預言到單晶制備再到實驗測量證實理論研究的另一個典范.
7 總 結
本文詳細介紹了最近幾年來幾種拓撲半金屬的單晶生長進展.從Dirac半金屬Na3Bi,Cd3As2和EuCd2As2,Weyl半金屬Mo/WTe2,Ir1?xPtxTe2和HgCr2Se4,Node-Line半金屬ZrSiS,TiB2,到“沙漏費米子”KHgSb和三重簡并費米子MoP,每種材料都對應著相應的生長方法.在常規條件下生長單晶主要用到助熔劑法和氣相傳輸法,這兩種方法適用于絕大部分材料的單晶生長.生長單晶的難點在于:一是如何選取合適的助熔劑以及傳輸介質,二是如何設定反應的溫度控制程序.通過研究樣品中各個元素的化學及物理特性,制定具有針對性的方案,能夠提高單晶生長的效率.
[1]Qi X L,Zhang S C 2011 Rev.Mod.Phys.83 1057
[2]Hasan M Z,Kane C L 2010 Rev.Mod.Phys.82 3045
[3]Weng H,Dai X,Fang Z 2016 J.Phys.:Condens.Matter 28 303001
[4]Wang Z J,Sun Y,Chen X Q,Franchini C,Xu G,Weng H M,Dai X,Fang Z 2012 Phys.Rev.B 85 195320
[5]Xu S Y,Liu C,Kushwaha S K,Sanar R,Krizan J W,Belopolski I,Neupane M,Bian G,Alidoust N,Chang T R,Jeng H T,Huang C Y,Tsai W F,Lin H,Shibayev P P,Chou F C,Cava R J,Hasan M Z 2015 Science 347 294
[6]Xiong J,Kushwaha S K,Liang T,Krizan J W,Hirschberger M,Wang W D,Cava R J,Ong N P 2015 Science 350 413
[7]Wang Z J,Weng H M,Wu Q S,Dai X,Fang Z 2013 Phys.Rev.B 88 125427
[8]Liang T,Gibson Q,Ali M N,Liu M H,Cava R J,Ong N P 2015 Nat.Mater.14 280
[9]Neupane M,Xu S Y,Sankar R,Alidoust N,Bian G,Liu C,Belopolski I,Chang T R,Jeng H T,Lin H,Bansil A,Chou F C,Hasan Z 2014 Nat.Commun.5 3786
[10]Weng H M,Fang C,Fang Z,Bernrviget B A,Dai X 2015 Phys.Rev.X 5 011029
[11]Lü B Q,Xu N,Weng H M,Ma J Z,Richard P,Huang X C,Zhao L X,Chen G F,Matt C E,Bisti F,Strocov V N,Mesot J,Fang Z,Dai X,Qian T,Shi M,Ding H 2015 Nat.Phys.11 724
[12]Huang X C,Zhao L X,Long Y J,Wang P P,Chen D,Yang Z H,Liang H,Xue M Q,Weng H M,Fang Z,Dai X,Chen G F 2015 Phys.Rev.X 5 031023
[13]Sun Y,Wu S C,Ali M N,Felser C,Yan B H 2015 Phys.Rev.B 92 161107
[14]Deng K,Wan G,Deng P,Zhang K N,Ding S J,Wang E Y,Yan M Z,Huang H Q,Zhang H Y,Xu Z L,Denlinger J,Fedorov A,Yang H T,Duan W H,Yao H,Wu Y,Fan S S,Zhang H J,Chen X,Zhou S Y 2016 Nat.Phys.12 1105
[15]Soluyanov A A,Gresch D,Wang Z J,Wu Q S,Troyer M,Dai X,Bernevig B A 2015 Nature 527 495
[16]Feng B,Chan Y H,Feng Y,Liu R Y,Chou M Y,Kuroda K,Yaji K,Harasawa A,Moras P,Barinov A,Malaeb W,Bareille C,Kondo T,Shin S,Komori F,Chiang T C,Shi Y G,Matsuda L 2016 Phys.Rev.B 94 195134
[17]Wu Y,Mou D X,Jo N H,Sun K W,Huang L N,Budko S L,Can fi eld P C,Kaminski A 2016 Phys.Rev.B 94 121113
[18]Xu Q N,Song Z D,Nie S M,Weng H M,Fang Z,Dai X 2015 Phys.Rev.B 92 205310
[19]Singha R,Pariari A K,Satpati B,Mandal P 2017 Proc.Natl.Acad.Sci.USA 114 2468
[20]Fu B B,Yi C J,Zhang T T,Caputo M,Gao X,Lü B Q,Kong L Y,Huang Y B,Shi M,Vladimir S,Fang C,Weng H M,Shi Y G,Qian T,Ding H 2017 arXiv:1712.00782[cond-mat.mtrl-sci]
[21]Chang T R,Chen P J,Bian G,Huang S M,Zheng H,Neupert T,Sankar R,Xu S Y,Belopolski I,Chang G Q,Wang B K,Chou F C,Bansil A,Jeng H T,Lin H,Hasan M Z 2016 Phys.Rev.B 93 245130
[22]Bian G,Chang T R,Sankar R,Xu S Y,Zheng H,Neupert T,Chin C K,Huang S M,Chang G Q,Belopolski I,Sanchez D S,Neupane M,Alidoust N,Liu C,Wang B K,Lee C C,Jeng H T,Zhang C L,Yuan Z J,Jia S,Bansil A,Chou F C,Lin H,Hasan M Z 2016 Nat.Commun.7 10556
[23]Guan S Y,Chen P J,Chu M W,Sankar R,Chou F C,Jeng H T,Chang C S,Chuang T M 2016 Sci.Adv.2 1600894
[24]Feng X,Yue C M,Song Z D,Wu Q S,Wen B 2018 Phys.Rev.Mater.2 014202
[25]Liu Z H,Lou R,Guo P J,Wang Q,Suan S S,Li C H,Thirupathaiah S,Fedorov A,Shen D W,Liu K,Lei H C,Wang S C 2017 arXiv:1712.03048[cond-mat.mtrl-sci][26]Wang Q,Guo P J,Sun S S,Li C H,Liu K,Lu Z Y,Lei H C 2017 Phys.Rev.B 97 205105
[27]Wang Z J,Alexandradinata A,Cava R J,Bernevig B A 2016 Nature 532 189
[28]Yan B,Müchler L,Felser C 2012 Phys.Rev.Lett.109 116406
[29]Ma J Z,Yi C J,Lü B Q,Wang Z J,Nie S M,Wang L,Kong L Y,Huang Y B,Richard P,Zhang P,Yaji K,Kuroda K,Shin S,Weng H M,Bernevig B A,Shi Y G,Ding H 2017 Sci.Adv.3 1602415
[30]Zhu Z M,Winkler G W,Wu Q S,Soluyanov A A 2016 Phys.Rev.X 6 031003
[31]Lü B Q,Feng Z L,Xu Q N,Gao X,Ma J Z,Kong L Y,Richard P,Huang Y B,Strocov V N,Fang C,Weng H M,Shi Y G,Qian T,Ding H 2017 Nature 546 627
[32]Ma J Z,He J B,Xu Y F,Lü B Q,Chen D,Zhu L W,Zhang S,Kong L Y,Gao X,Rong L Y,Huang Y B,Richard P,Xi C Y,Choi E S,Shao Y,Wang Y L,Gao H J,Dai X,Fang C,Weng H M,Chen G F,Qian T,Ding H 2018 Nat.Phys.14 349
[33]He J B,Chen D,Zhu W L,Zhang S,Zhao L X,Ren Z A,Chen G F 2017 Phys.Rev.B 95 195165
[34]Tachibana M 2017 Mechanisms of Crystal Growth from Fluxed Solutions.In:Beginner’s Guide to Flux Crystal Growth(Tokyo:Springer)pp23–41
[35]Bugaris D E,zur Loye H C 2012 Angew.Chem.Int.Ed.51 3780
[36]Wanklyn B M,Maqsood A 1979 J.Mater.Sci.14 1975
[37]Tachibana M 2017 Mechanisms of Crystal Growth from Fluxed Solutions.In:Beginner’s Guide to Flux Crystal Growth(Tokyo:Springer)pp61–74
[38]Yan J Q,Sales B C,Susner M A,McGuire M A 2017 Phys.Rev.Mater.1 023402
[39]Kaldis E 1974 Principles of the Vapour Growth of Single Crystals.In:Crystal Growth(Boston:Springer)pp49–191
[40]Schmidt P,Binnewies M,Glaum R 2013 Chemical Vapor Transport Reactions-Methods,Materials,Modeling.In:Advanced Topics on Crystal Growth(InTech Open)pp23–54
[41]Gruehn R,Glaum R 2000 Angew.Chem.Int.Ed.39 692
[42]Hua G,Nie S,Song Z,Yu R,Xu G,Yao K 2018 arXiv:1801.02806[cond-mat.mtrl-sci]
[43]Brauer G,Zintl E 1937 Zeitschrift für Physikalische Chemie 37 323
[44]Johnson C E,Fischer A K 1970 J.Less Common Metals 20 339
[45]Kushwaha S K,Krizan J W,Feldman B E,Gyenis A,Randeria M T,Xiong J,Xu S Y,Alidoust N,Belopolski I,Liang T,Hasan Z M,Ong N P,Yazdani A,Cava R J 2015 APL Mater.3 041504
[46]Gukov O Y,Ugai Y A,Pshestanchik V R,Goncharov E G,Pakhomova N V 1970 Phase Diagram of the System Cd–As.(USSR:Voronezh State Univ.)
[47]Gokcen N A(Massalski T B E ed.)1990 Binary Alloy Phase Diagrams(Vol.1)(2nd Ed.)(Materials Park Ohio:ASM International)pp320–323
[48]Palenzona A,Manfrinetti P,Fornasini M L 1998 J.Alloys Compd.280 211
[49]Lorenz R,Plumbridge D 1913 Zeitschrift für Anorganische und Allgemeine Chemie 83 228
[50]Fang A F,Xu G,Dong T,Zheng P,Wang N L 2013 Sci.Rep.3 1153
[51]Yan L,Zhao J Z,Li Y,Lin C T,Liang A J,Hu C,Ding Y,Xu Y,He S L,Zhao L,Liu G D,Dong X L,Zhang J,Chen C T,Xu Z Y,Weng H M,Dai X,Fang Z,Zhou X J 2015 Chin.Phys.Lett.32 067303
[52]Noh H J,Jeong J,Cho E J,Kim K,Min B I,Park B G 2017 Phys.Rev.Lett.119 016401
[53]Huang H,Zhou S,Duan W 2016 Phys.Rev.B 94 121117
[54]Zhang K,Yan M,Zhang H,Huang H,Arita M,Sun Z,Duan W,Wu Y,Zhou S 2017 Phys.Rev.B 96 125102
[55]Yan M,Huang H,Zhang K,Wang E,Yao W,Deng K,Wan G,Zhang H,Arita M,Yang H,Sun Z,Yao H,Wu Y,Fan S,Duan W,Zhou S 2017 Nat.Commun.8 257
[56]Fu B B,Yi C J,Wang Z J,Yang M,Lü B Q,Gao X,Li M,Huang Y,Fang C,Weng H M,Shi Y G,Qian T,Ding H 2017 arXiv:1712.02500[cond-mat.mtrl-sci]
[57]Fei F,Bo X,Wang P,Ying J,Chen B,Liu Q,Zhang Y,Sun Z,Qu F,Zhang Y,Li J,Song F,Wan X,Wang B,Wang G 2017 arXiv:1711.10909[cond-mat.mtrl-sci]
[58]Okamoto H(Massalski T B E Ed.)1990 Binary Alloy Phase Diagrams(Vol.3)(2nd Ed.)(Materials Park,Ohio:ASM International)pp3136–3139
[59]Okamoto H(Massalski T B E ed.)1990 Binary Alloy Phase Diagrams(Vol.3)(2nd Ed.)(Materials Park,Ohio:ASM International)pp2357,2358
[60]Lu H,Jia S 2017 Front Phys.12 127211
[61]Li Z L,Chen H X,Jin S F,Gan D,Wang W J,Guo L W,Chen X L 2016 Cryst.Growth Des.16 1172
[62]Xu G,Weng H M,Wang Z J,Dai X,Fang Z 2011 Phys.Rev.Lett.107 186806
[63]Wan X G,Turner A M,Vishwanath A,Savrasov S Y 2011 Phys.Rev.B 83 205101
[64]Wang Z J,Vergniory M G,Kushwaha S,Hirschberger M,Chulkove E V,Ernst A,Ong N P,Robert J C,Bernevig B A 2016 Phys.Rev.Lett.117 236401
[65]Brewer L,Lamoreaux R H(Massalski T B E ed)1990 Binary Alloy Phase Diagrams(Vol.3)(2nd Ed.)(Materials Park,Ohio:ASM International)pp2675,2676
[66]Ali M N,Xiong J,Flynn S,Quinn G,Leslie S,Haldolaarachchige N,Ong N P,Tao J,Cava R J 2014 Nature 514 205
[67]Okamoto H(Massalski T B E ed)1990 Binary Alloy Phase Diagrams(Vol.3)(2nd Ed.)(Materials Park,Ohio:ASM International)p3472
[68]Guan T,Lin C J,Yang C L,Shi Y G,Ren C,Li Y Q,Weng H M,Dai X,Fang Z,Yan S S,Xiong P 2015 Phys.Rev.Lett.115 087002
[69]Lin C J,Yi C J,Shi Y G,Zhang L,Zhang G M,Müller J,Li Y Q 2016 Phys.Rev.B 94 224404
[70]Lehmann H W,Emmenegger F P 1969 Solid State Commun.7 965
[71]Takahashi T 1970 J.Cryst.Growth 6 319
[72]Yu R,Weng H M,Fang Z,Dai X,Hu X 2015 Phys.Rev.Lett.115 036807
[73]Quintela C X,Campbell N,Shao D F,Irwin J,Harris D T,Xie L,Anderson,T J,Reiser N,Pan X Q,Tsymbal E Y,Rzchowski M S,Eom C B 2017 APL Matter.5 096103
[74]Schoop L M,Ali M N,Stra?er C,Topp A,Varykhalov A,Marchenko D,Duppel V,Parkin P S S,Lotsch B V,Ast C R 2016 Nat.Commun.7 11696
[75]Ali M N,Schoop L M,Garg C,Lippmann J M,Laa E,Lotsch B,Parkin P S S 2016 Sci.Adv.2 e1601742
[76]Higashi I,Takahashi Y,Atoda T 1976 J.Cryst.Growth 33 207
[77]Nakano K,Hayashi H,Imura T 1974 J.Cryst.Growth 24–25 679
[78]Sirtl E,Woerner L M 1972 J.Cryst.Growth 16 215
[79]Murray J L 1988 Metall.Trans.A 19 243
[80]Bradlyn B,Cano J,Wang Z J,Vergnioru M G,Felser V,Cava R J,Bernevig B A 2016 Science 353 6299
[81]Vogel R,Schuster H U 1980 Z.Naturforsch.35b 114
[82]Sangster J,Pelton A D 1993 J.Phase Equilib.14 510
[83]Kurnakow N S 1900 Z.Anorg.Allg.Chem.23 439
[84]Jangg G,Lihl F,Legler E 1962 Z.MetaIlkd.53 313
[85]Guerin R,Sergent M,Chaudron G 1975 CR Acad.Sci.Ser.C 281 777
[86]Shekhar C,Sun Y,Kumar N,Nicklas M,Manna K,Suess V,Young O,Leermakers I,Foerster T,Schmidt M,Muechler L,Werner P,Schnelle W,Zeitler U,Yan B H,Parkin S S P,Felser C 2017 arXiv:1703.03736[condmat.mtrl-sci]
[87]Olesinski R W,Abbaschian G J(Massalski T B E ed)1990 Binary Alloy Phase Diagrams(Vol.2)(2nd Ed.)(Materials Park,Ohio:ASM International)pp1967,1968
[88]Olesinski R W,Kanani N,Abbaschian G J(Massalski T B E ed)1990 Binary Alloy Phase Diagrams(Vol.2)(2nd Ed.)(Materials Park,Ohio:ASM International)pp 1978,1979
[89]Chi Z H,Chen X L,An C,Yang L X,Zhao J G,Feng Z L,Zhou Y,Zhou Y,Gu C C,Zhang B W,Yuan Y F,Kenney-Benson C,Yang W G,Wu G,Wan X G,Shi Y G,Yang X P,Yang Z R 2017 arXiv:1710.00472[condmat.supr-con]
猜你喜歡
祝您健康·文摘版(2024年6期)2024-07-26 00:00:00
小讀者(2021年2期)2021-03-29 05:03:48
少兒美術(2020年3期)2020-12-06 07:32:54
現代裝飾(2020年11期)2020-11-27 01:47:48
中學生天地(A版)(2020年3期)2020-04-10 10:57:45
故事作文·高年級(2020年3期)2020-03-17 09:24:33
瘋狂英語·新悅讀(2019年11期)2019-12-18 05:14:16
華人時刊(2019年13期)2019-11-17 14:59:54
NBA特刊(2018年21期)2018-11-24 02:48:04
文苑(2018年22期)2018-11-19 02:54:14
