液液萃取氣相色譜-ECD法同時測定飲用水中的13種農藥
2018-07-09 13:16:20劉慧杰張平允石金濤
凈水技術 2018年6期
劉慧杰,張平允,姜 蕾,石金濤,李 寧
(上海城市水資源開發利用國家工程中心有限公司,上海 200082)
目前還沒有測定環氧七氯的國標方法,《生活飲用水標準檢驗方法 農藥指標》(GB/T 5750.9—2006)分析六六六(BHC)、滴滴涕(DDT)、六氯苯、七氯等11種有機氯和溴氰菊酯農藥時,不同農藥的前處理過程、分析條件,甚至檢測儀器都不一致[1],按照GB/T 5750.9—2006的方法標準,11種農藥的富集需要經過4種不同的方法進行富集,檢測儀器涉及氣相、氣質聯用儀器以及液相色譜,分析過程費時費力、分析效率低。開發多組分農藥同時分析的方法成為近年來飲用水分析領域的熱點之一。譚美凌等[2]采用固相萃取的方法同時測定水中痕量的環氧七氯、七氯和六六六。液液萃取普及程度高,萃取效率高,文獻中報道的使用液液萃取氣相色譜法進行水體中有機氯及溴氰菊酯的方法研究已經有不少[3-5]。本文采用液液萃取氣相色譜法實現了環氧七氯、六六六、滴滴涕、六氯苯、七氯、溴氰菊酯13種農藥的同時測定。
本文采用正己烷作為萃取劑,一次萃取飲用水中13種農藥。通過優化氣相色譜的分析條件,大大縮短了分析時間,系統地考察了振蕩時間、鹽析劑加入量對回收率的影響,建立了一種簡便、快捷、準確、有效的飲用水中多種農藥同時檢測的氣相色譜方法。將該方法用于上海市飲用水出廠水檢測時,結果令人滿意。
1 試驗部分
1.1 主要儀器與試劑
安捷倫氣相色譜(Agilent 7890A)配有ECD檢測器;安捷倫自動進樣器(Agilent 7693);HP-5(30 m ×0.25 mm×0.25 μm)毛細管柱;濃縮儀:美國Caliper全自動樣品濃縮儀;正己烷為色譜純;環氧七氯、α-BHC、β-BHC、γ-BHC、δ-BHC、p,p′-DDE、p,p′-DDD、o,p′-DDT、p,p′-DDT、百菌清、七氯、六氯苯、溴氰菊酯13種農藥混標(100 μg/mL),均購自美國NSI公司(NSI Solution);無水硫酸鈉(分析純,國產,經500 ℃灼燒2 h后置干燥器內密封備用)。
1.2 樣品采集與保存
用棕色磨口玻璃瓶采集樣品,采集后的樣品于4 ℃的冰箱保存,并盡快完成分析。
1.3 樣品的前處理
用量筒量取100 mL水樣,加入到分液漏斗中,加入10 mL正己烷,置于自動振蕩器上,振搖時間為3 min;振蕩以后,靜置3~5 min直至其完全分層,棄去水相。正己烷萃取液通過玻璃三角漏斗(底部鋪有定性濾紙的無水硫酸鈉層)過濾,將萃取液收集于濃縮管中,用玻璃吸管移取正己烷淋洗無水硫酸鈉,使無水硫酸鈉完全潤洗,潤洗兩次,合并洗滌液于濃縮管中。將濃縮管放入濃縮儀中,于40 ℃水浴中濃縮至1.0 mL,搖勻。
1.4 標準溶液的配制
13種農藥標準工作溶液的配制:將13種農藥混標(1.1)稀釋為10 μg/mL,然后分別移取一定體積的上述標準溶液,稀釋為20、40、80、160、320、500 μg/L的13種農藥混合標準工作溶液。
1.5 氣相色譜分析條件
進樣口溫度為280 ℃;柱溫起始溫度為100 ℃,保持2 min,以25 ℃/min升至160 ℃,保持1 min,以15 ℃/min升至180 ℃,保持1 min,再以8 ℃/min升至260 ℃,保持3 min;載氣流速為1 mL/min;ECD檢測器加熱器:300 ℃,尾吹氣流量為30 mL/min;進樣量為1 μL不分流進樣。
1.6 測定及計算方法
以色譜峰的出峰順序和保留時間進行定性分析;以外標法進行定量,計算如式(1)。
(1)
其中:c1—水體中農藥化合物的濃度,μg/L;
c0—從標準工作曲線上查得的農藥化合物的濃度,μg/L;
V—水樣的體積,mL;
V1—萃取溶液的定容體積,mL。
2 結果與討論
2.1 色譜條件的優化
由于13種農藥沸點相差大,為使13種農藥進行良好的分離,采用程序升溫的方式,以減少組分的重疊和擴散。試驗發現,起始溫度低(60 ℃),保留時間大大延長,前18 min內,僅有溴氰菊酯出峰,整個分析時間為27 min,后12種物質出峰時間集中在20~27 min,影響檢測效率。若逐步提高程序升溫起始溫度,分析時間縮短。因此,試驗中采取提高起始溫度以縮短分析時間。但是起始溫度過高(大于150 ℃),各組分分離效果差,色譜峰有拖尾現象。綜合保留時間的長短與分離效果,本試驗采用(1.5)程序升溫方式進行程序升溫,13種農藥能夠完全分離,典型的分離色譜圖如圖1 所示。
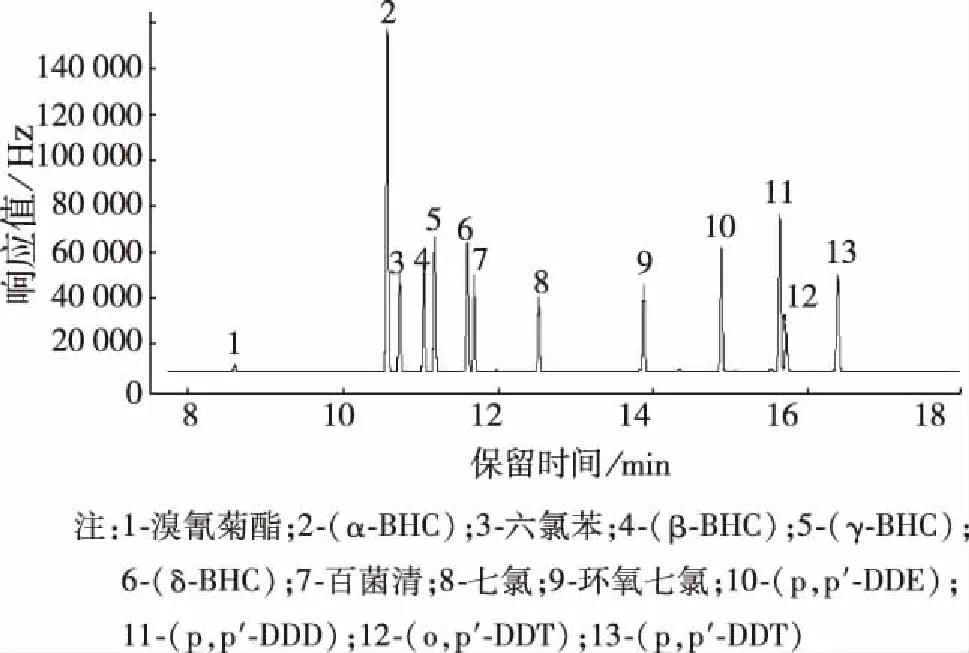
圖1 13種農藥化合物混合標準溶液氣相色譜圖(320 μg/L)Fig.1 Gas Chromatography of Mixed Standard Solutions for 13 Pesticides(320 μg/L)
2.2 萃取時間的優化
為優化萃取時間,向五份100 mL水中加標,使水中13種農藥的濃度為1 μg/L,分別振蕩3、5、8、10、15 min,各萃取液經儀器分析,平行測定2次,測定結果取均值,13種農藥的回收率如圖2所示。發現隨振蕩時間的增加,13種物質的回收率變化不大,其回收率均在90%~120%。因此,將萃取時間定為3 min。
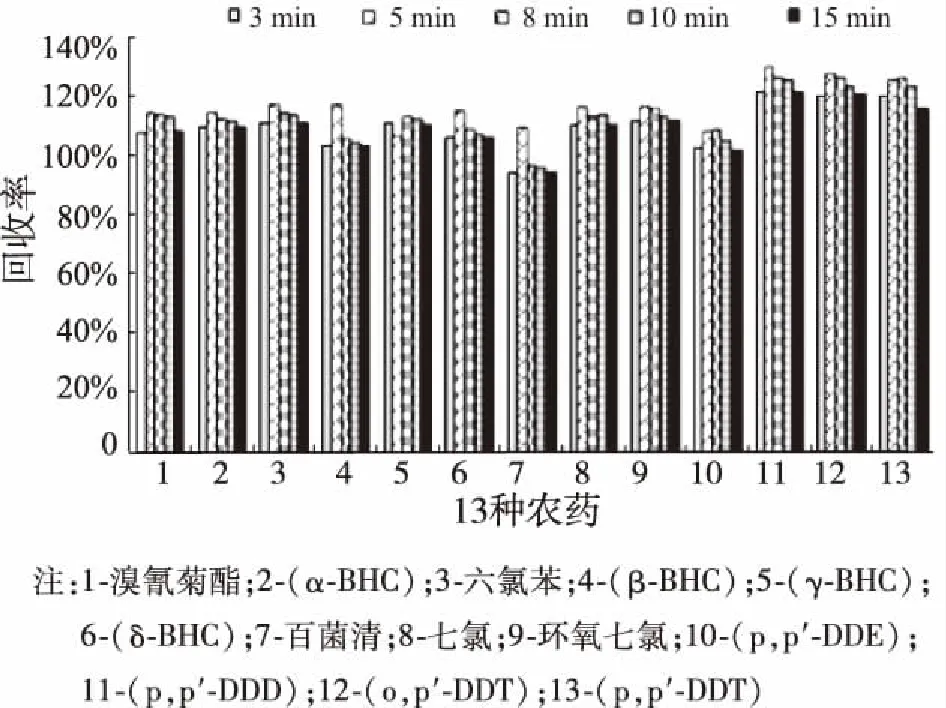
圖2 振蕩時間對回收率的影響(水樣中的目標物濃度為1 μg/L)Fig.2 Effect of Shaking Time on Recovery Rate(1 μg/L)
2.3 鹽析劑對萃取結果的影響
為考察鹽析劑濃度對萃取結果的影響,取五份高純水,依次分別加0、0.5、1.0、1.5、3.0 g氯化鈉,然后向五份高純水中加標,使其濃度為1 μg/L,平行測定2次,測定結果取均值,試驗結果如圖3所示。結果表明,在不加入氯化鈉的條件下,13種農藥的回收率均在96.9%~125%,隨著氯化鈉的加入量增加,13種農藥的回收率影響不明顯。說明,正己烷作為13種農藥的萃取試劑,萃取效果良好。因此,在萃取過程中,不加入鹽析劑。

圖3 氯化鈉投加量對回收率的影響(水樣中的目標物濃度為1 μg/L)Fig.3 Effect of Sodium Chloride Dosage on Recovery Rate(1 μg/L)
2.4 標準曲線及方法的檢出限
試驗用20、40、80、160、320、500 μg/L濃度梯度的混合農藥標準工作溶液 ,在1.5節的儀器條件下,以峰面積對濃度作校準曲線,經線性回歸得到各自線性回歸方程及相關系數,結果如表1所示。

表1 13種農藥化合物的線性方程與方法檢出限
由表1可知,各農藥化合物標準曲線線性方程良好,線性相關系數均大于0.995,均高于GB 5750.3—2006中對相關系數r>0.99的要求[6]。根據方法檢出限的確定方法[7],對濃度值為估計方法檢出限3~5倍的加標樣品進行n=7的平行測定。將各測定結果換算為樣品中的濃度(100 mL)水樣,計算平行測定的標準偏差,按照公式MDL=t(n-1,0.99)×S計算方法檢出限,式中n為樣品測定次數;自由度t為n-1,置信度為99 %時,t分布為單側,S為標準偏差。按照上述方法進行測定,13種農藥的方法檢出限如表1所示。各農藥化合物的檢出限均低于GB/T 5750.9—2006給出的方法檢出限,因此該方法滿足測定的要求。
2.5 實際水樣分析及加標回收率及精密度
采集14份上海市生活飲用水出廠水水樣,按照本文中分析方法,采用正己烷萃取,進行GC-ECD檢測。測定結果表明,采用該分析方法進行檢測,均未檢測出上述α-BHC、β-BHC、γ-BHC、δ-BHC、p,p′-DDE、p,p′-DDD、o,p′-DDT、p,p′-DDT、百菌清、七氯、六氯苯、溴氰菊酯及環氧七氯13種農藥。
選擇一份水樣,分成兩組,每組六份,向上述兩組水樣中加入13種農藥的標準溶液,配制農藥混合物質的濃度分別為0.3、3 μg/L,按照步驟1.3中樣品的萃取和分析過程分別測定6次,得到方法在不同濃度范圍內的相對標準偏差和回收率,結果如表2所示。由表2可知:在低濃度(0.3 μg/L)時,13種農藥的相對標準偏差為2.17%~7.67%,回收率為80.4%~131%;在高濃度(3 μg/L)時,13種農藥的相對標準偏差為1.02%~6.04 %,回收率為95.5 %~123 %。

表2 13種農藥化合物的相對標準偏差及平均回收率
3 結論
采用液液萃取方法對飲用水中13種農藥進行檢測,操作簡便,萃取時間短。通過改進氣相色譜條件,13種農藥能在18 min內完成分析,且分離效果良好。該分析方法能夠滿足13種農藥快速分析的要求,具有準確度高,精密度好,適用于飲用水行業水質檢測實際樣品的分析。
[1]生活飲用水標準檢驗方法 農藥指標:GB/T 5750.9—2006[S].
[2]譚美凌,張凌云,劉波.固相萃取/毛細管氣相色譜法測定水中痕量有機氯[J].中國給水排水,2010,26(20):129-131.
[3]江樹廣.氣相色譜法同時測六六六、滴滴涕、六氯苯、七氯、百菌清[J].中國給水排水,2011,27(18):92-95.
[4]徐虹,曹文婷,易承學,等.液液萃取氣相色譜法測定飲用水中百菌清等12 種農藥殘留[J].中國衛生檢驗雜志,2014,24(23):3363-3365.
[5]仇秀梅,董學林,劉亞東,等.液液萃取一氣相色譜法同時測定地下水中16種有機氯農藥[J].環境污染與防治,2016,38(11):72-78.
[6]生活飲用水標準檢驗方法 水質分析質量控制:GB/T 5750.3—2006[S].
[7]環境監測分析方法制修訂技術導則:HJ 168—2010[S].
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
兒童故事畫報(2019年5期)2019-05-26 14:26:14
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
