原發性中樞神經系統淋巴瘤1例報告
2018-07-04 11:44:52牛云梅
中風與神經疾病雜志 2018年6期
關鍵詞:信號
牛云梅, 吳 哲
原發性中樞神經系統淋巴瘤(primary central nervous system lymphoma,PCNSL)是指原發于顱內、眼、脊髓和軟腦膜等部位的非霍奇金淋巴瘤,并在明確診斷時,無中樞神經系統以外淋巴結受累。新診斷的 PCNSL 患者,如未經治療,中位生存期僅為2~3 個月[1]。臨床中該病罕見,表現缺乏特異性,易被誤診為瘤樣脫髓鞘病(tumefactive demyelinating lesions,TDLs)。 PCNSL病灶有自發消失的可能, PCNSl患者使用激素后可能會出現暫時性自發緩解,這些都為臨床診斷帶來了困難。現將我院收治的1例患者報道如下。
1 臨床資料
患者,女性,45歲,因“突發右側偏盲14個月,頭迷、頭痛10 d”于2014年5月20日入院。患者于2013年3月初無明顯誘因突發右側偏盲,無頭暈頭痛、惡心嘔吐等不適,曾于外院門診就診,行頭部MRI檢查示:丘腦及枕葉占位性病灶,有強化,外周水腫帶較重,未行手術及藥物治療,1 m后右側偏盲好轉,復查頭部MRI示:病灶明顯減少,半年后再次復查MRI示:病灶完全消失。入院前10 d患者無明顯誘因突發頭迷、頭痛,為脹痛伴搏動感,頭迷與體位無關,于當地醫院行頭部CT檢查示:顱內占位性病變(見圖1A),今為求進一步診治收入我科。專科查體:神志清楚,查體合作,言語正常,發音正常。雙瞳孔等大正圓,D≈3.0 mm,光反應靈敏。雙眼向各方向運動充分,無眼震。其余肌力、深淺感覺、指鼻試驗等未見異常。輔助檢驗:血、尿常規、肝腎功能、風濕系列、腫瘤系列、HIV、梅毒、肝炎等無異常。輔助檢查:視覺誘發電位(VEP):左眼刺激,正常。右眼刺激,P100潛伏期正常,P100波幅右枕較左枕降低。體感誘發電位(SEP)、腦干聽覺誘發電位(BAEP)正常。腰穿檢查:腦脊液呈淺黃色、壓力為180 mmH2O,腦脊液常規:總蛋白:PROT 1323 mg/L(120~600 mg/L),葡萄糖:GLU3.8 mmol/L(2.2~3.9 mmol/L),氯:CL-120 mmol/L(120~132 mmol/L),細胞數11×106/L(0~15),多核細胞NEU27%,單個核細胞LYM73%,涂片未見隱球菌,未見典型瘤細胞。頭部MR平掃+增強:右側額葉、胼胝體膝部、雙側顳葉及右側枕葉可見多發長T1、長T2信號,周圍伴大片狀水腫信號(見圖1B、C),增強后病灶呈結節狀及環形強化(見圖1D)診斷意見:腦內多發病變,脫髓鞘疾病?淋巴瘤?頭部MRS(3.0T):CHO明顯升高,CR、NAA明顯降低,呈典型腦腫瘤波形,診斷意見:腦內惡性腫瘤可能大。神經外科會診意見:建議行活檢取病理以明確診斷。患者及家屬均表示拒絕。我科考慮TDLs不除外,給予甲強龍500 mg沖擊及營養神經治療。治療后復查頭部MR平掃+增強示:病灶減少。患者自覺頭痛頭迷癥狀明顯好轉,于2014年6月10日出院。
出院后患者繼續口服激素減量治療,可正常生活。2017年12月26日患者因“左側肢體無力2 w,加重3 d”為主訴再次入院。入院2 w前患者出現左側肢體無力,走路向左偏斜,伴摔倒,3 d前左側肢體無力較前加重,1 d前出現嗜睡,為求進一步診治再次入我科。專科查體:神志嗜睡,言語查體無法配合,發音查體無法配合。雙瞳孔等大正圓,D≈3.0 mm,光反應遲鈍。Babinski征(L:+,R:+)。其余查體無法配合。輔助檢查:肝膽脾胰雙腎輸尿管膀胱超聲檢查:未見異常回聲,肺部CT平掃:雙肺及胸膜陳舊病變。右肺小結節。心包少量積液。頭部CT平掃(64排):右側丘腦可見團塊狀混雜密度影,周圍可見大片狀低密度灶水腫帶,右側側腦室受壓,中線結構左偏,鄰近腦溝裂變窄。左側腦室增寬加深(見圖1E)診斷意見:右側丘腦占位性病變,建議MR增強檢查。頭部MR平掃+增強:右側丘腦可見不規則形混雜信號,范圍約5.6 cm×3.8 cm×5.0 cm(左右×前后×上下),T1WI以稍低信號為主(見圖1F),T2WI以稍高信號為主(見圖1G),增強后可見明顯強化(見圖1H),周圍腦實質可見水腫信號,增強后未見強化,右側腦室受壓、變形,局部中心結構左移;右側腦橋、小腦腳、延髓、雙側腦室旁白質可見斑片狀長T2信號,FLAIR序列高信號,幕上腦室系統擴張,腦溝池裂明顯增寬,診斷意見:右側丘腦占位性病變,膠質瘤?頭部DWI:右側丘腦團塊狀信號影周邊腦室旁及基底節區可見條片狀彌散受限高信號,診斷意見:右側丘腦占位性病變伴瘤周血管源性水腫改變,請結合臨床及增強MR檢查結果。頭部MRS(3.0T):單體素感興趣區位于右側丘腦,Cho峰增高,NAA峰減低;多體素病灶感興趣區位于左額葉,基線不穩,病變Cho波峰升高,NAA波峰降低,部分Lac峰倒置,Lip峰增高,Ch/NAA范圍1.71~2.9,Ch/Cr范圍1.52~3.34,診斷意見:右側腦內病灶MRS改變,符合低度膠質瘤可能大,請結合臨床。給予患者甲強龍1.0 g沖擊治療及丙球治療后,患者狀態不見好轉。后患者轉至神經外科在全麻下行大腦開顱右側丘腦腫瘤切除術,術后患者家屬放棄治療,回家后隨即死亡。
最終術后病理回報(見圖2):病理診斷:右側頂葉:免疫組化結果符合非霍奇金B細胞淋巴瘤(彌漫大B細胞淋巴瘤、活化B細胞型)免疫組化:A1:CK(-),CD3(-),CD20(+),Pax-5(+),CD10(-),Bcl-6(+)MUM1(+),CD30(+),Ki-67(+80%),Vimentin(+),GFAP(-),Olig2(-),NeuN(-),Synaptophysin(-),P53(),CD34(血管+),IDH1(+)。
2 討 論
PCNSL是比較罕見的淋巴瘤類型。病理上大部分為彌漫性大B細胞淋巴瘤(DLBCL,diffuse large B-cell lymphoma),其余的為少見的T細胞淋巴瘤、Burkitt淋巴瘤、間變性大細胞淋巴瘤等[2]。由于CNS中不存在淋巴組織,因此PCNSL的確切發病機制尚不明確。PCNSL常見發病部位為大腦半球(尤其額葉、顳葉)深部腦白質、丘腦/基底節區、胼胝體和腦室周圍,少數累及后顱窩、軟腦膜[3]。本例患者為中年女性,慢性起病,既往體健,無器官移植、HIV病史, 患者2013年因出現偏盲行頭部MRI檢查,發現丘腦及枕葉病灶,未行任何治療,6 m后MRI顯示病灶自行消失。2014年因出現頭迷、頭痛住院就診, MRI顯示腦內多發病灶,呈結節樣強化,MRS提示符合腦腫瘤表現。建議患者行腦活檢被拒, 給予激素治療后患者癥狀好轉出院。出院后患者因未出現明顯不適癥狀未定期復查頭部MRI,于2017年再次住院。此次入院影像學檢查呈丘腦占位,強化明顯。行活檢后病理診斷PCNSL明確。回顧患者長達5 y的病史。2013年病灶未經治療自行消失,2014年按TDLs給予激素治療后癥狀好轉,2017年再發,激素及丙球治療無效,術后病理為DLBCL,綜合患者全部病程考慮,2013年發病仍為PCNSL。
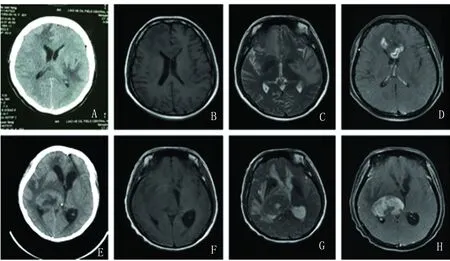
圖1 2014年頭部CT顯示:右側額葉、胼胝體膝部、雙側顳葉及右側枕葉可見低密度影(圖A)磁共振顯示:右側額葉、胼胝體膝部、雙側顳葉及右側枕葉可見多發長T1(圖B)、長T2信號(圖C),周圍伴大片狀水腫信號,增強后病灶呈結節狀及環形強化(圖D),2017年頭部CT顯示:右側丘腦可見團塊狀混雜密度影(圖E),磁共振顯示右側丘腦可見不規則形混雜信號,范圍約5.6×3.8×5.0 cm(左右×前后×上下),T1WI以稍低信號為主(圖F),T2WI以稍高信號為主(圖G),增強后可見明顯強化(圖H)
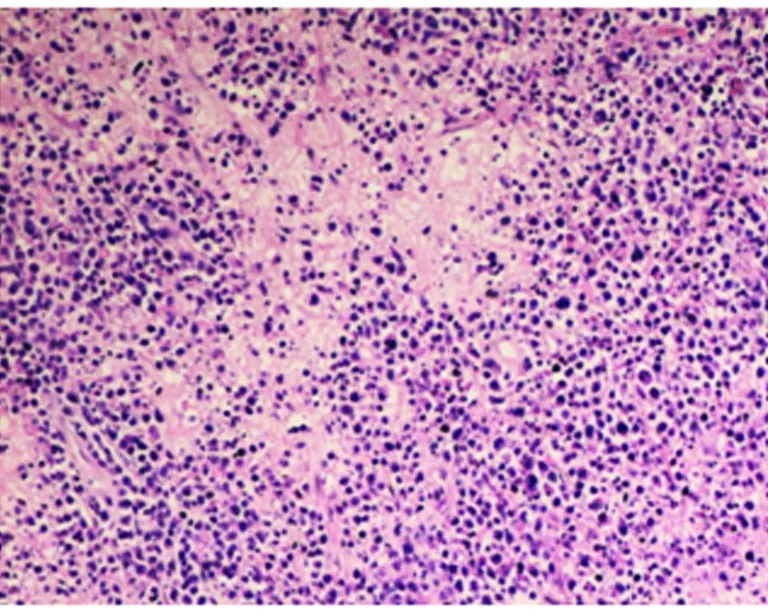
圖2 光鏡可見:瘤細胞呈彌散分布,細胞體積大,核大深染,核漿比例失調,核分裂象易見,異型性明顯
患者病史有其特殊性,即曾經歷過病灶自發消失,1 y后再發,使用激素后臨床癥狀及影像學緩解,緩解期長達3.5 y。檢索國內外文獻,我們發現了一些類似病例。國內劉靜等[6]2018年曾報道1例29歲女性,首次發病以“視物模糊、視野缺損”為主要臨床表現,激素沖擊治療后病灶逐漸消失,間隔2 y后再次發病經活檢才證實為PCNSL。檢索PubMed報道的病例中有3例類似。Keisuke TAYA等[7]2004年報道了一位主訴為“頭痛”的60歲女性患者, MRI顯示小腦有明顯增強的病灶,2001年2月14日,患者接受探查性開顱手術并部分切除腫瘤。2001年2月23日進行術后MRI顯示病灶消失。病理診斷為:PCNSL。隨訪MRI,術后1 y,MRI顯示小腦再次出現病灶。Steven等[8]2006年報道的病例為29歲的產后婦女,患者最初于2001年因“頭痛,嗜睡6 w,左側面癱和肢體無力4 d”入院。 頭部MRI顯示左側基底節和內囊后肢附近存在增強病灶。給予地塞米松口服有效。1 w后,腦病灶活檢顯示炎性脫髓鞘。隨后3 y,她一直很健康,隨訪的MRI顯示病灶幾乎完全消失。2005年2月產后出現局灶性癲癇發作、輕偏癱和顱內壓增高。MRI顯示右側額葉有兩個增強病灶,18F-FDG PET顯示高代謝。盡管再次大劑量激素沖擊治療,癥狀仍然復發。再次腦活檢證實為PCNSL。Sasaki等[9]2015年報道了1例既往體健的55歲女性患者。7 y前(2008年),患者出現左面部感覺減退。頭部MRI顯示右側丘腦病灶。但是感覺減退和顱內病變在6 m內自發消失。1 y前(2014年),患者出現右側肢體的運動障礙,頭部MRI顯示雙側基底節和左內囊的病灶逐漸增大。此前,她的左眼罹患過兩次激素敏感性葡萄膜炎。最初醫生懷疑是一種炎癥性疾病,如多發性硬化。激素治療雖然改善了她的臨床癥狀并減少了病灶的大小,但是在1 m內腦部病灶和雙側視神經炎復發。后經活檢證實為PCNSL。作者認為葡萄膜炎和視神經炎是由與PCNSL相關的眼內淋巴瘤引起的。患者2014年頭部MRI表現出皮質多發長T1、長T2的病灶,伴有強化。激素沖擊后復查:影像學原病灶緩解、臨床癥狀緩解。3.5 y后出現右側丘腦強化的淋巴瘤病灶。這在PCNSL中并不少見。PCNSL顱內可有炎性浸潤表現,可早于或并發于淋巴瘤實質病灶出現,并與腫瘤病灶位置不同。這一現象被稱為“前哨病變”,是PCNSL的一種罕見表現,其臨床表現及激素治療效果可與中樞神經系統脫髓鞘(如多發性硬化)非常相似,腫瘤實質病灶出現前常難以鑒別。“前哨病變”可以自發或使用糖皮質激素后自行消失,并且通常在隨后6~12 m內診斷PCNSL[14]。一部分學者認為這些炎癥反應是免疫系統對腫瘤的應答表現,而另一些學者認為淋巴瘤是炎性病變后單克隆細胞惡變的結果[13]。既往文獻報道的24例伴有前哨病變的PCNSL患者中,前哨病變發展至確診PCNSL的時間平均為20.1 m(4~65 m),病程多具有波動性。糖皮質激素治療通常可使臨床或影像緩解(95.8%)[4],其主要作用機制不是減輕病灶周圍的水腫,而是通過細胞毒作用導致淋巴瘤細胞凋亡[5]。該例患者發病早期病灶呈浸潤性,再次發病時呈實性占位性,提示兩種不同病灶可能是腫瘤不同發病階段的表現。劉靜等[6]認為淋巴瘤的影像學表現與其病理學特點密切相關。PCNSL沿血管周圍間隙浸潤性生長早期首先形成多發性衛星病灶,此時影像學表現多為浸潤性病灶。隨著腫瘤細胞的不斷增殖最后融合為實體瘤,此時影像學則表現為實性占位性病變。患者2014年入院時只有顱內壓增高癥狀,影像學顯示為顱內占位性病變,激素治療有效,容易誤診為TDLs。TDLs[10]是中樞神經系統一種相對特殊類型的免疫介導的炎性脫髓鞘病變。既往也稱瘤樣炎性脫髓鞘病(tumor like inflammatory demyelinating disease,TIDD),或脫髓鞘假瘤(demyelinating pseudotumor,DPT)。TDLs無典型的臨床表現,影像所見病變體積較大,多伴周邊水腫,具有占位效應,易與PCNSL相混淆。孫辰婧等[11]對13例TDLs與PCNSL互相誤診的病例進行回顧性分析,發現如下特點:(1)PCNSL的起病年齡顯著大于TDLs,前者主要見于中老年人群,而TDLs則以青中年居多;(2)TDLs的頭部CT病灶無高密度[12],而PCNSL多為高密度,若表現為低密度或等密度需與TDLs相鑒別;(3)在頭部MRI增強中,TDL頭部MRI可見動態演變,急性期為點狀或斑片狀強化,亞急性期為半環或環樣強化,慢性期強化程度慢慢減低;PCNSL頭部增強MRI呈均勻團塊樣強化。
PCNSL無典型的臨床癥狀,容易誤診為TDLs,“前哨兵變”有自發或使用激素后消失可能,盡管PCNSL患者可能會出現暫時性自發緩解,但我們目前的病例表明,從緩解到復發的時間間隔可能比一般預期的要長得多,緩解期可達數年之久。由此,報道本例希望可以為臨床醫生診治此類疾病提供幫助。
[參考文獻]
[1]Kalus S,Di MB,Gaillard F.Demyelination preceding a diagnosis of central nervous system lymphoma[J].Journal of Clinical Neuroscience Official Journal of the Neurosurgical Society of Australasia,2016,24:146-148.
[2]張劍寧,程 崗.原發性中樞神經系統淋巴瘤的再認識[J].中華神經外科疾病研究雜志,2016,15(1):1-4.
[3]Giannini C,Dogan Ahmet,Salomo DR.CNS lymphoma A practical diagnostic approach[J].Journal of Neuropathology and Experimental Neurology,2014,73(6):478-94.
[4]張包靜子,高名士,樊 潔,等.以進行性腦萎縮及非占位性腦白質病變起病的原發性中樞神經系統淋巴瘤(附1例報道及文獻復習)[J].中國臨床神經科學,2017,25(4):404-410.
[5]Weller M.Glucocorticoid treatment of primary CNS lymphoma[J].Journal of Neuro-Oncology,1999,43(3):237-239.
[6]劉 靜,魏清柱,呂田明,等.原發性中樞神經系統淋巴瘤發病不同階段影像學和病理學特點[J].中國現代神經疾病雜志,2018,18(01):41-46
[7]Taya K,Terao T,Tanaka T,et al.Spontaneous regression of primary central nervous system lymphoma:a case report[J].脳神経外科,2004,32(6):637-642.
[8]Ng S,Butzkueven H,Kalnins R,et al.Prolonged interval between sentinel pseudotumoral demyelination and development of primary CNS lymphoma[J].Journal of Clinical Neuroscience,2007,14(11):1126-1129.
[9]Sasaki T,Nakayama T,Kitamura M,et al.Case of 55-year-old female with primary central nervous system lymphoma,presenting with brain and eye lesions and long-term relapsing/remitting course[J].Clinical Neurology,2015,55(8):567.
[10]劉建國,胡學強.中樞神經系統瘤樣脫髓鞘病變診治指南[J].中國神經免疫學和神經病學雜志,2017,24(5):305-317.
[11]孫辰婧,洪 柳,劉建國,等.顱內腫瘤樣脫髓鞘病與原發性中樞神經系統淋巴瘤臨床誤診病理分析[J].中華神經科雜志,2015,48(9):757-762.
[12]劉建國,董秦雯,張海玲,等.病理證實的瘤樣脫髓鞘病60例影像學特點[J].中華神經科雜志,2014,47(10):680-686.
[13]Husseini L,Saleh A,Reifenberger G,et al.Inflammatory demyelinating brain lesions heralding primary CNS lymphoma[J].Canadian Journal of Neurological Sciences Le Journal Canadien Des Sciences Neurologiques,2012,39(1):6-10.
[14]Alderson L,Fetell MR,Sisti M,et al.Sentinel lesions of primary CNS lymphoma[J].J Neurol Neurosurg Psychiatry,1996,60(1):102-105.
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
