AARS2基因突變相關的腦白質營養不良1例并文獻分析
2018-07-04 11:44:50劉方方張康慶劉亢丁
中風與神經疾病雜志 2018年6期
關鍵詞:基因突變
劉方方, 張康慶, 劉亢丁, 鄧 方
腦白質營養不良是一組髓鞘形成或維持發生障礙的遺傳性疾病,遺傳因素決定其異質性[1]。遲發性白質腦病(Late-onset leukoencephalopathy),是一種常染色隱性遺傳的神經退行性疾病,屬于AARS2基因突變相關的腦白質營養不良,主要表現為進行性共濟失調、痙攣、認知功能的下降伴額葉功能障礙。腦磁共振顯示明顯的腦部深部白質改變和小腦萎縮,女性患者常伴有卵巢功能的衰竭[2]。文中收集1例相對少見的遲發性白質腦病的臨床、影像和實驗室檢查資料,并結合復習國內外文獻進行分析,希望加深臨床醫生對遲發性白質腦病的認識。
1 臨床資料
患者,男,34歲,因進行性走路不穩、肢體抖動7 y入院。患者于7 y前開始出現不自主雙手抖動,多于靜止時發作,持續半小時后自行緩解,行腦電圖等檢查排除癲癇發作,未治療;此后于緊張時出現雙手及雙下肢抖動。4 y前逐漸出現左下肢發沉乏力,左手力弱,尚可完成日常生活及工作。2 y前出現左上肢輕度屈曲,行走時踩棉花感,走路不穩,常向右側偏斜。同時出現下頜左右陣性擺動。約1.5 y前患者出現小便失禁,不愿參加社交活動。半年前患者無法獨立行走及坐起,臥床,并有二便失禁。并出現陣發性四肢抽動,主要表現為雙上肢屈曲強直,雙下肢伸直抖動,持續10 min左右,夜間抽動頻繁,可達3~4次。期間意識清楚,問話可答。父母非近親結婚,無類似家族史。否認煙、酒史,否認毒物、藥物接觸史。圍產期平穩。抬頭、走路均較同齡兒晚,智能等表現基本正常。服兵役期間不能耐受長途跑步訓練。神經系統檢查:癡笑面容,認知、記憶力及計算力差,構音障礙。雙側瞳孔等大同圓,直徑約3.0 mm,雙側直接、間接對光反射靈敏,雙眼上視略受限,其余眼球運動尚可,雙眼會聚查體不合作。舌體輕微震顫,無舌肌萎縮,伸舌略向右偏。咽反射消失,軟腭上抬受限。右肢肌力4+級,左上肢肘關節攣縮,左下肢肌力查體不合作,刺激肢體可見明顯躲避動作。雙下肢肌張力鉛管樣增高。右側腱反射活躍。雙側病理征陽性,左側踝陣攣陽性,余查體不配合。頭部MRI示雙側腦白質改變及腦萎縮,隨時間漸行加重(見圖1)。血清、腦脊液化驗及影像學檢查基本除外自身免疫性疾病、副腫瘤相關性疾病、內分泌代謝性疾病及感染性疾病。因嬰幼兒期發育遲緩,抽取患者及其父母外周血5 ml進行全外顯子測序。患者樣本在AARS2基因外顯子區域發現兩處雜合突變:c.1871G>A(鳥嘌呤>腺嘌呤),導致氨基酸改變p.W624*(色氨酸>終止);c.452T>C(胸腺嘧啶>胞嘧啶),導致氨基酸改變p.M151T(甲硫氨酸>蘇氨酸)。患者父母分別攜帶單一雜合突變(見圖2)。根據相關文獻報道最終確診為遲發性白質腦病。
2 討 論
AARS2基因定位于6號染色體,負責編碼線粒體丙氨酰t-RNA合成酶。AARS2基因突變影響線粒體內蛋白質合成,氧化磷酸化系統缺陷,進而引起多系統功能障礙[3]。線粒體通過合成蛋白質為呼吸鏈氧化磷酸化系統提供關鍵亞基[4]。線粒體物質代謝與生物學功能受核基因和線粒體基因共同調控。氧化磷酸化復合體約有80多種蛋白質,其中只有13種由線粒體基因組編碼,其余的均由核基因組編碼,在胞質核糖體合成后運輸到線粒體[5]。線粒體涉及的遺傳方式包括母系遺傳和孟德爾遺傳。由于受精卵的mtDNA來源于卵母細胞,故mtDNA的遺傳方式為母系遺傳。與線粒體病相關的nDNA編碼的基因遺傳方式屬于孟德爾遺傳,包括常染色體隱性遺傳、顯性遺傳和X-連鎖遺傳[6]。2011年首次發表了AARS2突變和人類疾病有關,其中有3例患有嬰兒線粒體心肌病的患者存在該基因的常染色體隱性突變[7]。后來Dallabona等描述了6例伴卵巢功能衰竭的腦白質營養不良患者經外顯子測序均存在AARS2突變[3]。Hamatani等于2016年首次報道1例亞洲AARS2突變引起的腦白質營養不良[8]。2016年David 等人研究中發現5例成人發病白質腦病伴軸突球和色素沉著癥患者攜帶AARS2的復合雜合或純合突變[9]。
AARS2基因突變可導致兩種不同表型的疾病:嬰兒線粒體心肌病和遲發性白質腦病[10]。2011年Gotz等通過外顯子測序證實了AARS2基因突變可引起嚴重的先天性肥厚性心肌病,這些患兒幾乎全部合并線粒體呼吸鏈缺陷的心肌病[7]。值得注意的是,2014年Dallabona等報道6例由于AARS2基因突變引起的伴有卵巢功能衰竭的白質腦病患者,但并沒有心肌病的跡象[3]。2015年,Euro等對這一現象做出解釋。 AARS2基因共包含22個外顯子,負責編碼線粒體氨基酰-tRNA合成酶除了有氨酰化區域外,還有一個編輯校正區域,負責水解錯誤連接的tRNA與氨基酸,另外還有一個C-末端,負責提高tRNA與氨基酸結合及氨酰化的速率,各亞區共同作用從而保證線粒體蛋白合成的高效及精確性[7]。研究發現心肌病患者常存在編輯校正區域突變位點,而腦白質營養不良患者并沒有出現此區域突變位點。因此,心肌病患者氨酰化缺陷程度較腦白質營養不良患者的缺陷程度重[4]。
AARS2相關性白質腦病需要與其他腦白質病變相鑒別,特別是與成年發病的白質腦病合并軸索球樣變和顆粒樣膠質細胞(ALSP)。ALSP是遺傳性彌漫性白質腦病合并軸索球樣變(HDLS)的一種類型,為一種罕見的常染色體顯性遺傳腦白質病,突變基因為集落刺激因子1受體基因(CSFl R),定位于5號染色體,其突變主要影響單核巨噬細胞、神經膠質細胞的增殖和分化[8]。兩者均可表現為額葉頂端的腦室周圍白質疾病伴皮質下不對稱的U型纖維節段性病變,錐體束和胼胝體均可受累。但ALSP可見明顯中央萎縮和胼胝體變薄,而AARS2相關性白質腦病中則相對輕微且不成比例;ALSP偶爾也會在額葉表現出腦室周圍腦白質異常和鈣化,但在AARS2相關性白質腦病中不存在鈣化改變。此外與ALSP不同的是,AARS2相關性白質腦病的女性病例常見卵巢萎縮[11]。
在本文中我們描述了一名中年男性患者,慢性進展性起病,病程中未見明顯緩解過程。與外界交流少,認知、記憶力及計算力差,高級神經功能受累。舌體震顫、肢體抖動、肌張力增高,錐體外系受累。雙側病理征陽性,錐體束受累。抽搐時家屬可動其肢體,大汗,家屬訴晨間勃起減少,遺精減少,考慮自主神經功能受累。患者坐立不能不除外與小腦受累所致平衡功能障礙及背部肌肉肌張力增高有關。基因檢測發現患者母親存在一處雜合突變:c.1871G>A(鳥嘌呤>腺嘌呤);其父親存在一處雜合突變:c.452T>C(胸腺嘧啶>胞嘧啶);患者為重合雜合突變,存在c.1871G>A和c.452T>C兩處突變。研究發現此兩個位點均有較低的人群攜帶率,其中c.1871G>A為終止突變,對蛋白功能的影響可能較大。此雙雜合突變分別來自于其父母,為復合雜合突變,符合常染色體隱性遺傳規律。上述兩個突變點為首次發現,推測其均有致病性,但目前尚無與這兩個突變點有關的文獻報道,故需相應的純合突變病例來進一步證實。
根據患者的基因檢測結果及相關文獻復習,AARS2基因的突變與其臨床表現相符合,可以很好的解釋其癥狀的發生、發展與轉歸。迄今為止,包括本案在內涉及AARS2突變有關遲發性白質腦病大部分都是散發性的,沒有家族史,并且是由復合雜合突變引起的,使得診斷非常困難。臨床上磁共振示腦白質多發異常信號應警惕遲發性白質腦病的可能,必要時進行基因篩查。該患的基因發現可能為AARS2基因相關的遲發性白質腦病提供進一步的證據并擴大其突變譜。
[參考文獻]
[1]Kaye EM,Moser H.Where has all the white matter gone? Unraveling the mysteries of leukoencephalopathies[J].Neurology,2004,62(9):1464-1465.
[2]鄭 揚,章殷希,丁美萍.卵巢性腦白質營養不良的研究進展[J].中華神經科雜志,2017,50(5):384-387.
[3]Dallabona C,Diodato D,Kevelam SH,et al.Novel (ovario) leukodystrophy related to AARS2 mutations[J].Neurology,2014,82(23):2063-2071.
[4]Euro L,Konovalova S,Asin-Cayuela J,et al.Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation[J].Front Genet,2015,6:21.
[5]Scheffler IE.A century of mitochondrial research:achievements and perspectives[J].Mitochondrion,2001,1(1):3-31.
[6]劉 譽,韋建鴿,吳彬彬,等.線粒體病的分子生物學機制[J].暨南大學學報(自然科學與醫學版),2011,32(2):115-121.
[7]Gotz A,Tyynismaa H,Euro L,et al.Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy[J].Am J Hum Genet,2011,88(5):635-642.
[8]Hamatani M,Jingami N,Tsurusaki Y,et al.The first Japanese case of leukodystrophy with ovarian failure arising from novel compound heterozygous AARS2 mutations[J].J Hum Genet,2016,61(10):899-902.
[9]Lynch DS,Zhang WJ,Lakshmanan R,et al.Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia[J].JAMA Neurol,2016,73(12):1433-1439.
[10]Szpisjak L,Zsindely N,Engelhardt JI,et al.Novel AARS2 gene mutation producing leukodystrophy:a case report[J].J Hum Genet,2017,62(2):329-333.
[11]Lakshmanan R,Adams ME,Lynch DS,et al.Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy[J].Neurol Genet,2017,3(2):135.

圖1 患者頭部MRI改變。圖a~d為患者2012年頭部MRI改變;圖e~h為2018年頭部MRI改變。圖示:患者雙側基底節、放射冠、側腦室旁、半卵圓中心、額頂葉、胼胝體膝部及壓部見斑點狀及斑片狀異常信號,改變隨時間漸加重,可見腦溝腦裂加深,腦組織萎縮漸明顯
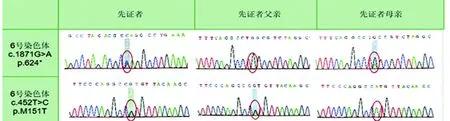
圖2 患者家系成員Sanger測序結果。先證者其母攜帶AARS2基因chr6:44272054位點c.1871G>A雜合突變,其父攜帶AARS2基因chr6:44279256位點c.452T>C的雜合突變,先證者為AARS2基因復合雜合突變
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
