伴眼部異常的TTR型FAP一家系報告及文獻復習
2018-07-04 11:44:50胡波琳盧曉慶漆學良丁衛江
中風與神經疾病雜志 2018年6期
胡波琳, 盧曉慶, 漆學良, 丁衛江
家族性淀粉樣多神經病變(familial amyloid polyneuropathy,FAP)是一種罕見的常染色體顯性遺傳病,成人發病,淀粉樣物質沉積于全身多個系統,以周圍神經系統受累最為明顯,診斷的金標準是基因檢測。目前研究認為FAP主要是由轉甲狀腺素蛋白(transthyretin,TTR)、載脂蛋白A-1(apolipoprotein A-1,Apo-A1)和凝溶膠蛋白的變異引起。其中TTR相關FAP(TTR-FAP)最常見,且病情最嚴重,常危及生命。1984年首次發現該病的致病基因[1]。此后經過研究,目前已發現100多個突變位點[2]。TTR-FAP可累及眼部,主要表現為玻璃體混濁。不過由于對該疾病認識的不足,病理活檢的有創性及基因診斷的未廣泛普及,目前國內報道的家系較少,推測低于實際發病率,故現將我院收治的1個伴有視物模糊的TTR型FAP家系臨床資料和基因檢測結果報道如下。
1 資料與方法
患者,男性,33歲,主因“雙下肢無力3 m余”就診于我院。主要表現為走平路和上樓梯時抬腿吃力感,休息數分鐘后緩解。夜間偶爾全身肌肉有針刺感和肉跳。既往否認高血壓、糖尿病。因視物模糊發現雙玻璃體混濁,已行手術治療。家族中其祖父、父親、1個叔叔、2個姑姑、2個堂兄弟均有肢體無力及視物模糊,其家系圖譜如下(見圖1)。患者的堂哥(Ⅲ3)出現四肢無力,雙眼玻璃體混濁,對稱性非梗阻型肥厚型心肌病;患者的另一個堂哥(Ⅲ1)表現為肢體無力,伴玻璃體出血。入院查體:神志清楚,高級皮質活動正常,雙眼球向各方向活動自如,雙眼瞼閉合有力,雙眼視力:右眼0.5,左眼:0.6。雙側面部及肢體痛觸覺對稱正常,咽反射對稱存在,雙下肢近端肌力4-級,遠端肌力4+級,雙上肢肌力5-級。四肢腱反射減低,雙下肢痛溫覺減退,雙下肢肌萎縮明顯。病理征陰性,腦膜刺激征陰性。共濟運動存在。完善相關檢查:血常規:WBC 9.75×109/L(正常值3.5~9.5×109/L),血小板計數124×109/L(正常值125~350×109/L),尿常規、糞便常規、凝血4項、血清葡萄糖、肝腎功能、電解質、乙肝6項、輸血4項均未見異常。我院肌電圖(EMG)顯示廣泛性周圍神經損害:(1)運動神經遠端潛伏期延長、傳導速度減慢;雙下肢運動神經復合動作電位波幅衰減;感覺神經電位波幅低平,感覺傳導速度減慢。(2)正中神經、脛神經刺激所獲F波潛伏期延長,波形離散。(3)針級肌電圖示較多正相、纖顫電位,輕收縮時雙下肢肌肉運動單位電位平均時限增寬,波幅增高。大力收縮時運動單位電位數量減少,呈單純相。心電圖示竇性心率,部分導聯ST-T段抬高。胸部平片未見明顯異常X線征象。雙眼及附屬器B超示:雙眼玻璃體混濁。雙側眼底照相顯示:雙眼底出血。雙眼OCT示:右眼屈光介質混濁,OCT信號弱,隱約見黃斑區網膜結構大致正常;左眼屈光介質混濁,OCT無信號,未見黃斑區網膜結構。完善右側腓腸神經病理活檢結果示:有髓神經纖維重度缺失伴剛果紅陽性淀粉樣物質沉積。電鏡結果:電鏡下膠原纖維增多,有髓神經纖維數目明顯減少,部分髓鞘極度松散,神經束膜下見成片聚合物,由長度不等,相互交錯排列,淀粉樣物質聚合而成,超微結構觀察顯示:淀粉樣變,神經缺失。Ⅲ3、先證者(Ⅲ5)及IV2完善基因檢查(送檢至北京康旭醫學檢驗所),結果顯示:Ⅲ3及先證者TTR基因均出現c.220G>A雜合突變(見圖2);Ⅳ2基因結果未見該突變。支持該患者TTR-FAP的診斷。

圖1 患者家系圖譜
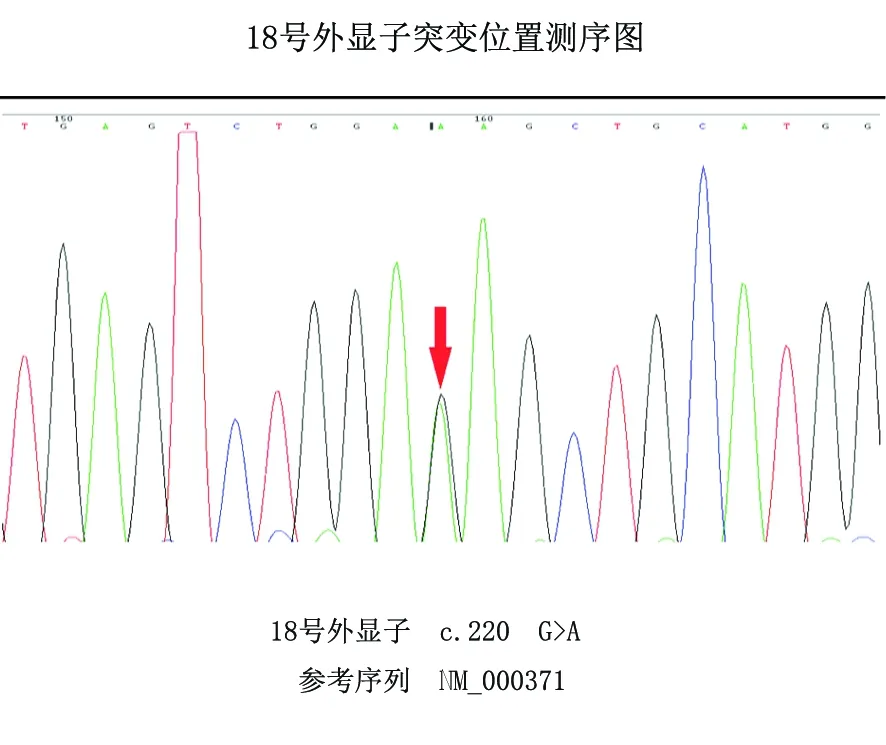
圖2 先證者(III10)TTR基因檢測結果顯示18號外顯子突變
2 討 論
TTR-FAP診斷標準:(1)臨床診斷:①明確的陽性家族史;②早期發生并且隨著病程逐漸進展的多神經病癥狀;③電生理檢查發現大小神經纖維的損害;(2)病病理診斷:剛果紅染色發現淀粉樣蛋白沉積,免疫組織化學染色顯示為TTR淀粉樣蛋白;(3)基因診斷:發現特定的TTR基因突變。其中基因診斷為診斷該疾病的金標準[3]。基于上述診斷標準。結合患者的病史、體征、家族史、病理學檢查、神經電生理檢查及基因檢測,該患者有充分的依據診斷為TTR-FAP。
TTR-FAP最常見的眼部病變為玻璃體混濁,混濁物為黃白色棉絨樣團塊狀或片狀物質,隨時間的延長逐漸增多[4~6]。眼部B型超聲檢查顯示玻璃體團塊狀回聲混濁[7]。眼部異常表現為玻璃體出血的罕見。2007年OHeam TM等報道了2例玻璃體積血患者發生在一個罕見的TTR突變glu54gly中[8],此后TTR-FAP伴玻璃體出血的報道少。在該家系中3人均表現出明顯的眼部異常:其中2人伴明顯玻璃體混濁;1人伴玻璃體出血。研究發現TTR 可由脈絡叢及視網膜色素上皮細胞合成,致病性突變降低了TTR四聚體的穩定性,促進其解離成單體,導致單體在胞外自行聚集,進一步形成寡聚體和淀粉樣蛋白,最終發生玻璃體混濁[9]。玻璃體積血形成的原因可能是淀粉樣蛋白在視網膜血管壁沉積,阻礙了周圍組織的氧輸送,缺氧后誘導血管內皮生長因子表達上調;也可能繼發于淀粉樣浸潤所致的血管壁損傷[10]。現于這一家系中發現伴玻璃體出血患者,推測可能是該突變基因所導致的淀粉樣蛋白易于沉積于視網膜血管壁,導致最后的出血。該家系中共3個患者出現明顯的眼部異常,1個出現玻璃體出血,發生玻璃體出血的概率為(1/3),發生率高于目前的報道。推測可能是因為少量或者微量玻璃體出血被明顯的玻璃體混濁掩蓋了,導致玻璃體出血目前報道的發生率低于實際發生率。但玻璃體混濁和玻璃體出血可能也是同一種疾病的不同表現形式。隨訪半年來未再發現玻璃體出血患者,仍需要繼續隨訪。
TTR-FAP是由TTR基因突變導致的常染色體顯性遺傳病[11],具有基因型和表型異質性[12]。在1984年首次發現該病的突變基因:Val30Met,同時該突變也是目前世紀范圍內該病的最常見的突變類型[13]。不過基因的突變仍存在一定的地域差異:在歐洲和日本發病率最高的是Val30Met[14,15]。目前國內報道的與淀粉樣變有關的突變有Leu55Arg、Lys35Asn、Ser100Ser、Lys35Thr、Gly83Arg、Arg54Gly等[16]。大多數的TTR-FAP是TTR基因雜合突變所致。在該家系中,出現的也是TTR基因的雜合突變:c.220G>A,即編碼區第220號核苷酸由 G 變為 A的雜合核苷酸變異,該變異導致第 74 號氨基酸由 Glu 變為 Lys(p.Glu74Lys)。該突變在人群中發生的頻率極低,國外該變異導致的淀粉樣多發性神經病已經文獻報道[17],而目前中國還未見該突變致病性的報道。這一家系中通過基因檢測明確致病的突變為Glu74Lys,雖然目前發現引起的玻璃體淀粉樣變性的突變位點眾多,但這是目前國內首次發現該突變基因,這一發現增加了我國該疾病的突變基因譜,為將來的該疾病的研究增加了臨床數據。并且TTR-FAP患者的外顯率并不一定是百分之百,研究發現該疾病在流行區域外顯率明顯增加:在葡萄牙,Val30Met的外顯率達87%,而在瑞典僅為2%[18]。在該家系中有3人完善基因檢測,出現基因突變的患者均出現明顯的臨床癥狀,外顯率為100%。該家系來自中國南方,推測中國南方可能為該疾病的高發區。
TTR-FAP 的治療相對困難。因為TTR主要在肝臟合成,肝移植是目前最有效的治療,研究發現肝移植術后患者血清中的變異TTR持續快速下降,病情趨于穩定。至于藥物治療,口服氯苯唑酸可以防止蛋白錯誤折疊和突變的TTR沉積,已經被批準在歐盟上市[19]。基因療法及治療性單克隆抗體尚處于研究階段但是目前未見肝移植及藥物治療對眼部病變有效性的報道。玻璃體完全切除術是目前治療該病的有效方法,可顯著提高患者的視力,但若切除不完全易導致復發。與此同時,研究發現全視網膜激光光凝可阻止淀粉樣物質在玻璃體和視網膜表面沉積。
[參考文獻]
[1]Saraiva MJ,Birken S,Costa PP,et al.Amyloid fibril protein in familial amyloidotic polyneuropathy,Portuguese type.Definition of molecular abnormality in transthyretin (prealbumin)[J].J Clin Invest,1984,74(1):104-119.
[2]Benson MD,Kincaid JC.The molecular biology and clinical features of amyloid neuropathy[J].Muscle Nerve,2007,36(4):411-423.
[3]Adams D,Suhr OB,Hund E,et al.First European consensus for diagnosis,management,and treatment of transthyretin familial amyloid polyneuropathy[J].Curr Opin Neurol,2016,29(1):S14-26.
[4]Zou X,Dong F,Zhang S,et al.Transthyretin Ala36Pro mutation in a Chinese pedigree of familial transthyretin amyloidosis with elevated vitreous and serum vascular endothelial growth factor[J].Exp Eye Res,2013,110:44-49.
[5]Liu T,Zhang B,Jin X,et al.Ophthalmic manifestations in a Chinese family with familial amyloid polyneuropathy due to a TTR Gly83Arg mutation[J].Eye(Lond),2014,28(1):26-33.
[6]Kimura A,Ando E,Fukushima M,et al.Secondary glaucoma in patients with familial amyloidotic polyneuropathy[J].Arch Ophthalmol,2003,121(3):351-356.
[7]林海燕,李 瑩,杜 虹.家族性淀粉樣變多發性神經病變伴雙眼多發病變1例[J].中華眼科雜志,2014,50(10):790-791.
[8]OHeam TM,Fawzi A,He S,et al.Early onset vitreous amyloidosis in familial amyloidotic polyneuropathy with a transthyretin Glu54Gly mutation is associated with elevated vitreous VEGF[J].Br J Ophthalmol,2007,91(12):1607-1609.
[9]Merlini G,Bellotti V.Molecular mechanisms of amyloidosis[J].N Engl J Med,2003,349(6):583-596.
[10]Dub EJ,Yang HS,Hailer JA,et al.Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor: implications for ocular angiogenesis[J].Am J Ophthalmol,2004,137(4):668-674.
[11]Ando Y,Coelho T,Berk JL,et al.Guideline of transthyretin-related hereditary amyloidosis for clinicians[J].Orphanet J Rare Dis,2013,8:31.
[12]Adams D,Lozeron P,Lacroix C.Amyloid neuropathies[J].Curr Opin Neurol,2012,25(5):564-572.
[13]Plante Bordeneuve V,Said G.Familial amyloid polyneuropathy[J].Lancet Neurol,2011,10(12):1086-1097.
[14]Zaros C,Genin E,Hellman U,et al.On the origin of the transthyretin Val30Met familial amyloid polyneuropathy[J].Ann Hum Genet,2008,72(4):478-484.
[15]Olsson M,Jonasson J,Cederquist K,et al.Frequency of the transthyretin Val30Met mutation in the northern Swedish population[J].Amyloid,2014,21(1):18-20.
[16]王輝林,龍 達,曾 軍,等.家族性淀粉樣多神經病致病基因突變分析[C].全國醫學遺傳學學術會議,2009.
[17]謝 兵,蔡善君,蔣 模,等.家族性玻璃體淀粉樣變性甲狀腺激素結合蛋白Gly83Arg突變家系[J].中華眼底病雜志,2016,32(3):89-91.
[18]Shi Y,Li J,Hu J,et al.A new Arg54Gly transthyretin gene mutation associated with vitreous amyloidosis in Chinese[J].Eye Sci,2011,26(4):230-238.
[19]Misu Ki,Hattori N,Nagamatsu M,et al.Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan.Clinicopathological and genetic features[J].Brain,1999,122(10):1951-1962.
