NQO1在tBHQ干預急性CO中毒后遲發性腦病大鼠海馬區的表達及作用
2018-06-21 09:14:44張亦雯張益梅李經倫
中風與神經疾病雜志 2018年5期
吳 沙,呂 霞,何 林,張亦雯,張益梅,李經倫
含碳物質的不完全燃燒生成過量一氧化碳(Carbon monoxide,CO),人體吸入過量CO后引起的中毒稱急性一氧化碳中毒(Acute carbon monoxide poisoning,ACOP),是臨床工作中常見的急危重癥病,致死致殘率極高。在生活生產中,通氣不良的環境下以煤氣中毒、炭火燃燒、工業泄漏等形式威脅著人們的生活。急性一氧化碳中毒遲發性腦病(delayed encephalophathy after acute carbon monoxide poisoning,DEACMP)即神經精神后發癥,是指約10%~30%急性CO中毒患者早期即使經成功救治意識障礙恢復后,仍表現出在一定時間的“偽恢復期”(2~60 d)后出現以精神心理行為異常、癡呆癥狀,以及錐體系、錐體外系癥狀為主的神經系統癥狀和體征[1]。因其高致死率、高致殘率無疑已成為ACOP患者最為嚴重的并發癥,其發病幾率在重度CO中毒中可達10%~40%,預后不佳,給社會和家庭帶來嚴重的經濟負擔,同時多年來該病的預防及治療也成為擺在國內外無數臨床醫師面前的極大挑戰。目前對于該病發病機制的研究蕓蕓眾多,十分復雜,但仍無統一定論,且任意一種均不能齊全地解釋其具體發病特點。近幾年來核因子2-相關因子2/抗氧化反應元件(nuclear factor erythroid 2 p45-related factor 2/anti-oxidant response element,Nrf2/ARE)通路在抗氧化應激中發揮著舉足輕重的作用,而ACOP中毒的本質為缺氧引發的一系列氧化應激反應,近年來研究表明該通路在DEACMP發病中發揮著重要作用,醌氧化還原酶1(NAD(P)H:quinone oxidoreductase 1,NQO1)作為其下游經典靶基因之一,是調節細胞內物質處于氧化還原狀態的黃素酶[2],因其催化醌類物質無單電子中間產物及自由基產生而發揮保護細胞作用已為人們所熟知,但隨著研究的深入其弊端也逐漸浮現。該研究通過在成功建立ACOP中毒后遲發性腦病(DEACMP)大鼠模型,運用Nrf2激動劑叔丁基對苯二酚(tertiary buty lhydroquinone,tBHQ)干預大鼠觀察其下游靶基因NQO1的表達進一步探討NQO1在該病發病中發揮的作用,為進一步研究DEACMP 發病機制提供實驗參考,致力于減少臨床上DEACMP 的發生,為探究其靶向治療奠定基石、提供指導。
1 材料與方法
1.1 主要的實驗材料
1.1.1 實驗動物 在成都達碩實驗動物有限公司購買健康的清潔級SD雄性大鼠180只,飼養于西南醫科大學城北動物實驗中心,體重約300 g,生產許可證號:SCXK(川)2016~15。在溫度20~25 ℃、濕度約40%~70%、光暗12 h交替環境、給予充足的鼠糧及高壓消毒純凈水飼養;
1.1.2 主要實驗試劑 在瀘州市西南化工研究院購買CO純品氣體(中國),南京華立明化工購買tBHQ試劑(中國),圣克魯斯生物技術公司的兔抗鼠NQO1多克隆抗體(中國),Roche公司的Western熒光試劑、Tunel試劑盒(德國),Dako公司Envision試劑盒(丹麥)。
1.1.3 主要實驗儀器 購自中國成都泰盟軟件有限公司的Morris水迷宮實驗儀、美國雅培公司的血氣分析儀、英國BRIGHT公司的石蠟切片機、美國徠博公司的倒置顯微鏡、美國ProteinSimple公司的凝膠成像儀。
1.2 實驗方法
1.2.1 實驗分組 模型建立、干預處理:將飼養并訓練1 w的雄性SD大鼠進行Morris水迷宮實驗,去掉有學習記憶功能缺失的大鼠,從180只可取的大鼠中隨機抽出60只作為空氣對照組(AC組),剩余大鼠用于建立遲發性腦病模型,按染毒后1 d、3 d、7 d、14 d、21 d、28 d分為6個亞組,染毒方式如下[3]:首劑CO以100 ml/kg體重腹腔注射,每隔4 h以50 ml/kg體重劑量追加注射,連續追加3次。AC組予以同等方式注射空氣。腹腔注射CO的大鼠隨機分成TC組60只、CO組60只,按50 mg/kg體重每日一次相同時間點腹腔注射tBHQ溶劑于TC組大鼠,CO組大鼠不做其他處理。
1.2.2 模型判定 動脈血碳氧血紅蛋白(HbCO)濃度測定:從TC、CO組每個亞組中隨機抽取1只大鼠,AC組中隨機抽取1只,按染毒前及注射首劑CO后的不同時間點(15 min,2 h,4 h,6 h,8 h,10 h,12 h,14 h,16 h,20 h)采集0.5 ml股動脈血,血氣分析儀測HbCO濃度。采用Morris水迷宮實驗方法驗證大鼠學習記憶能力的變化。
1.2.3 取材、標本處理 從各組中按每個時間點隨機選出一部分大鼠,適量生理鹽水進行左心室心臟灌洗,4%多聚甲醛灌注固定后斷頭取腦,4%多聚甲醛中保存,48 h內冠狀位切取含海馬、齒狀回的腦組織進行石蠟包埋、切片。
1.2.4 免疫組織化學法檢測NQO1的表達 烤片脫蠟、梯度酒精水化,PBS沖洗,內源性過氧化物酶失活,枸櫞酸納緩沖液抗原修復,加一抗室溫1 h,4 ℃過夜,37 ℃復溫45 min,PBS沖洗后加二抗,辣根過氧化物酶酶標,DAB顯色3~10 min,顯微鏡下調控染色水平,蘇木精復染,脫水透明封片,鏡下觀察,采用人工半定量計數法對陽性表達細胞進行統計分析[4]。
1.2.5 Western Blot檢測NQO1 蛋白 提取蛋白,配膠,加樣,變性離心,放入4 ℃冰箱備用,上樣、電泳、轉膜、封閉后進行免疫反應、顯色,經凝膠成像儀成像,以B-actin為參考值,得出NQO1條帶密度相對值,與內參值比較得出目標條帶灰度值,進行統計分析。
1.2.6 TUNEL染色 梯度酒精脫蠟,滴加試劑酶標、DAB 顯色,復染后脫水透明、封片,40×10倍光鏡下觀察海馬CA1區細胞凋亡情況,算出凋亡指數進行統計分析。
2 結 果
2.1 染毒后行為學表現 與AC組比較,TC組、CO組大鼠染毒后不久(<10 min)即有躁動不安、過度呼吸表現,CO追加次數增多后發生精神萎靡、四肢乏力、呼吸困難、末梢循環缺氧表現,部分大鼠甚至發生肢體強直抽搐、大小便失禁、喪失意識、死亡(8只)。保持通風良好的空氣環境中完成操作,及時補充相應數目大鼠的同時將死亡的大鼠剖腹解剖,其內臟不同程度腫脹、顏色暗紅、表面散在出血點,剝開顱骨發現其腦組織彌漫性充血水腫。其余存活大鼠攝食減少、精神萎靡、易受驚嚇。Morris水迷宮實驗結果顯示中毒大鼠隨染毒時間延長其活動路徑以隨機、自由、散漫形式多見,毫無規律。染毒后14 d始平均潛伏期較AC組明顯延長、平臺象限活動時間、穿越平臺頻次明顯減低(P<0.05),直至28 d,TC組上述變化明顯較CO組顯著(P<0.05)。
2.2 動脈血HbCO濃度變化 染毒前各組大鼠動脈血HbCO濃度均<1%,首劑注射CO 15 min后,HbCO濃度攀升達65%左右,2 h時仍高于60%,4 h時降至55%左右,此時進行追加半劑CO后測HbCO濃度繼續攀升至70%左右,而后下降走勢,整個染毒過程大鼠體內HbCO濃度>50%持續超過16 h。
2.3 海馬區NQO1的表達
2.3.1 免疫組織化學結果 AC組大鼠海馬CA1區NQO1蛋白表達極少,各時間點無明顯差異。TC組、CO組大鼠海馬CA1區NQO1蛋白的陽性細胞在40×10倍顯微鏡下胞核和胞質出現深淺不一的棕褐色或棕黃色,呈上升-峰點(3 d)-下降的走勢,TC組峰點較CO組峰點更高更明顯,TC組、CO組組內染毒后3 d與其他時間點比較及各組間各相同時間點比較差異具有統計學意義(見圖1,見表1)。
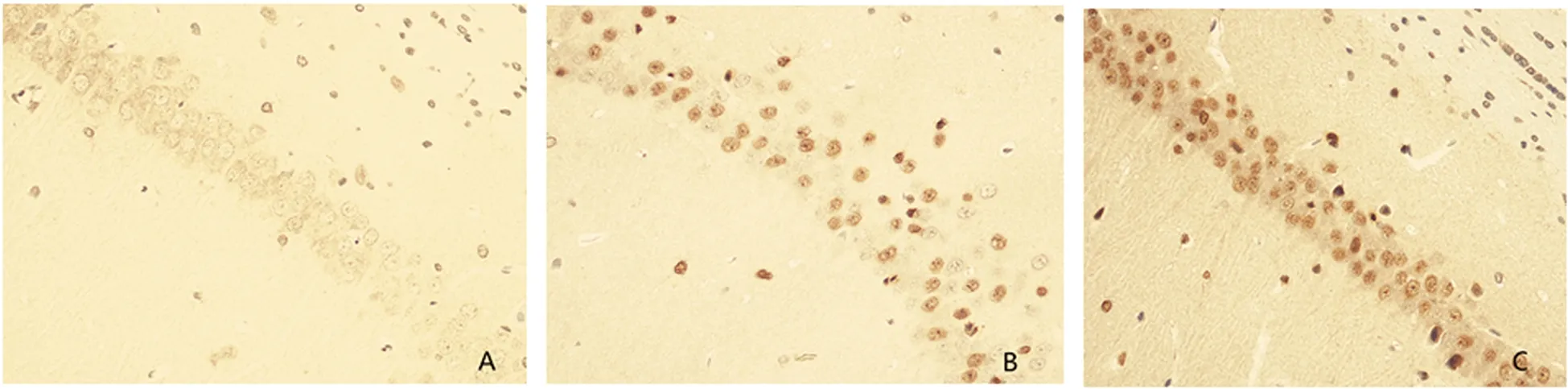
圖1 A、B、C分別為染毒后3 d,AC組、CO組、TC組大鼠海馬CA1區NQO1蛋白表達情況,陽性細胞為棕褐色或棕黃色
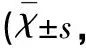
表1 3組大鼠海馬CA1區不同時間點NQO1免疫組化陽性結果比較分)
注:與AC組比較各相同時間點*P<0.05;與CO組比較△P<0.05;組內:TC組內▽P<0.05;CO組內▲P<0.05
2.3.2 Western Blot結果 AC組大鼠海馬NQO1蛋白表達不顯著,CO組、TC組大鼠海馬中NQO1的表達(見圖2)。
2.4 Tunel染色 AC組大鼠凋亡細胞少,各時間點無明顯變化。TC組、CO組大鼠染毒后均出現不同程度的細胞凋亡現象,從1 d開始逐步攀升,7 d達峰點,而后逐漸下降,且組內峰期凋亡情況與其他時間點比較差異具有統計學意義。達峰點(7 d)前TC組較CO組細胞凋亡指數下降,達峰點之后TC組較CO組細胞凋亡指數升高,除14 d外其他各時間點組間比較差異具有統計學意義(見圖3,表2)。
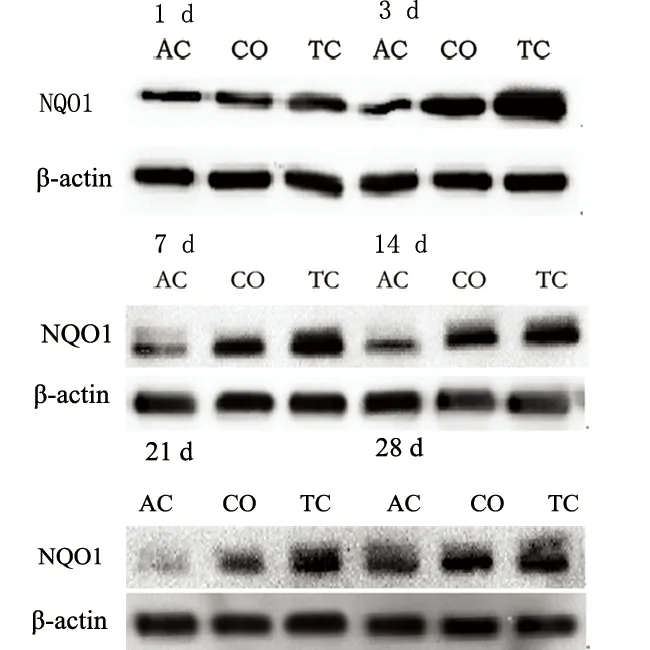
圖2 Western Blot結果顯示NQO1的表達條帶
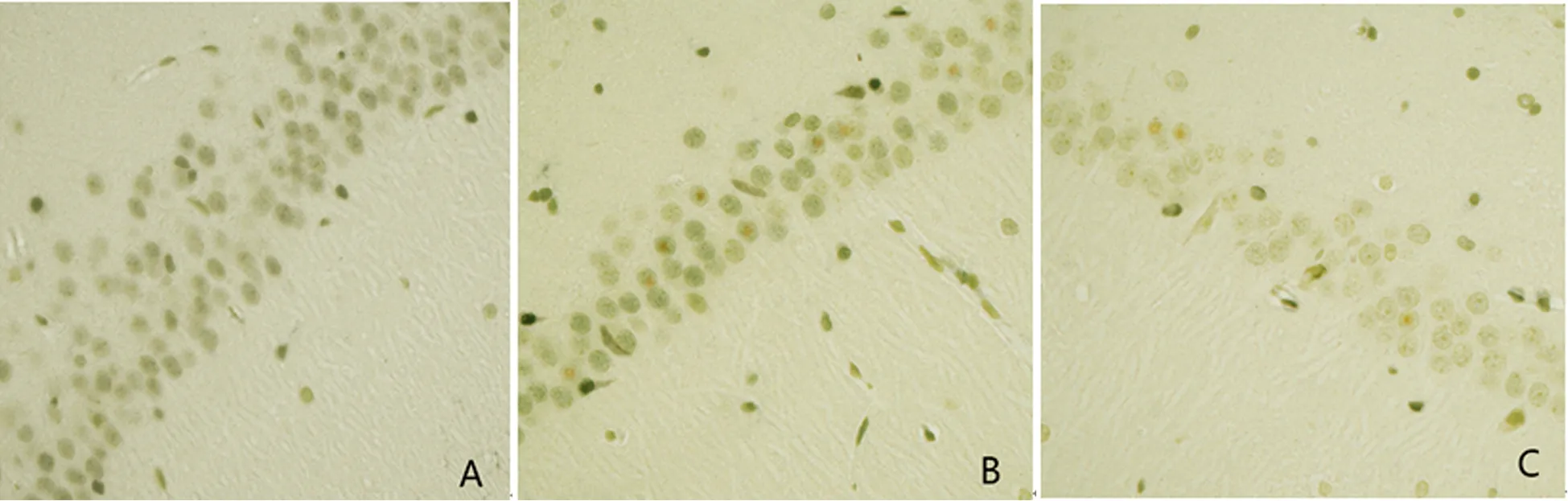
圖3 A、B、C分別為染毒后7 d的AC組、CO組、TC組大鼠海馬CA1區細胞凋亡情況,胞內含棕色顆粒的細胞為凋亡細胞
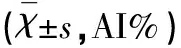
表2 3組大鼠海馬CA1區不同時間點細胞凋亡指數的比較
注:與AC組比較各相同時間點*P<0.05;與CO組比較△P<0.05;TC組內▽P<0.05,CO組內▲P<0.05
3 討 論
NQO1(醌氧化還原酶1、DT-diaphorase)是真核生物細胞內廣泛存在的一種可誘導的黃素蛋白酶,最早由Ernster 和Navazio于1958年報道[5]。胞質中多見,由NADH或NADPH提供電子催化醌類及其衍生物發生雙電子還原反應,催化反應過程中無單電子還原產物或自由基生成故能保護細胞免受氧化應激損傷[6],可被缺氧環境、抗氧化劑、多環芳烴、偶氮染料等誘導表達,這種可誘導性使得NQO1在腫瘤預防及治療上發揮舉足輕重的作用,缺氧和高氧化應激是腫瘤微環境的標志性特征之一,研究發現NQO1在消化、呼吸、生殖、皮膚系統等多種腫瘤性疾病中高表達提示預后不良,患者生存率降低[7,8]。DEACMP實質是缺氧誘發氧化應激反應后導致的一系列腦功能障礙疾病,抗氧化劑還可通過生物異源性物質反應元件(XRE)激活 NQO1基因的表達,NQO1主要存在于細胞質中,以NADH或NADPH作為電子供體,催化包括醌、醌亞胺、亞甲藍、二氯靛酚、谷胱甘肽取代的蔡醌、偶氮和硝基化合物等等在內的物質發生還原反應,不生成親電子還原產物及自由基,可解毒醌為毒性較低的氫醌類化合物而清除,避免對細胞的損傷,是公認的解毒防癌酶[9]。NQO1參與維生素E介導的抗氧化,充當輔酶Q的還原酶,維持P53蛋白穩定從而起到保護神經元細胞免受氧化應激以及一系列其他內源性毒素損傷的效果[10];在慢性神經系統變性疾病如:帕金森病和肌萎縮側索硬化中誘導NQO1的表達上調能夠起到很好的神經保護作用[11],阿爾茨海默病(AD)患者腦組織中大量神經細胞丟失、凋亡,神經細胞外老年斑沉積,神經纖維纏結,其中就富含NQO1[12]。且有動物實驗結果發現[13]NQO1基因敲除小鼠腦出血發生率及神經細胞損傷嚴重程度顯著高于野生型小鼠。
而近年來有研究發現,經NQO1還原而生成的產物因其結構及穩定性差異而可能對細胞產生相應的毒副作用[14]。經NQO1催化生成的產物氫醌對細胞凋亡影響存在量效、時效關系,當氫醌在細胞內達一定濃度或持續作用一定時間后不僅誘導細胞凋亡還引起細胞繼發性壞死[15]。其機制可能如下:(1)低劑量氫醌可能誘導HSP、peroxiredoxin等保護性蛋白表達以使細胞發生適應性反應保護細胞,但細胞氧化應激反應激烈時,cys發生不可逆的活化,上述保護性蛋白發生過氧化而失活,細胞內氧化還原失衡最終致細胞死亡[16]。(2)氫醌通過激活內源性細胞凋亡途徑,促發Caspase級聯反應,尤其是Caspase-3、Caspase-7活化,誘導細胞程序化死亡[17,18]。(3)氫醌可激活蛋白激酶B,使其定位于胞質中調控下游信號分子FasL、P27、cyclinE、cyclinB等表達影響細胞周期及發生凋亡作用[19,20]。(4)氫醌可經多途徑代謝,代謝過程可產生活性氧(ROS),ROS通過直接參與細胞信號通路,使線粒體膜電位變化促發線粒體細胞凋亡程序[21]。(5)氫醌可激活DNA-PKcs/Akt信號通路,活化Akt(蛋白激酶B)進一步推動氧化應激反應,氧化應激正反饋促進Akt活化,增強細胞對ROS誘發細胞凋亡的作用[22]。
tBHQ為廣泛應用于人們生活生產中的抗氧化劑,同時是受Nrf2/ARE調節的二相解毒酶誘導劑,能調節該通路下游靶基因NQO1、HO-1、GSH等的表達[23],有實驗發現20 μmol/LtBHQ處理細胞,胞核中Nrf2在加藥后8 h表達最佳,其下游靶蛋白NQO1在胞質中加藥后16 h表達最佳,撤去tBHQ后其誘導表達持續4 h[24]。故本實驗采用染毒后每日一次相同時間點腹腔注射經乙醇溶解的濃度為5 mg/mL的tBHQ,達到其持續誘導NQO1表達的效果。另外本實驗采用成熟的改良式腹腔注射CO法建立ACOP中毒遲發性腦病大鼠模型,使大鼠體內動脈血HbCO濃度>50%至少持續達16 h之久,能達到類似臨床中重度CO中毒患者的程度,更利于DEACMP發病[25]。
Morris水迷宮實驗發現TC、CO組大鼠在染毒后前7 d并未出現智能障礙,這與臨床DEACMP患者發病經歷潛伏期相似。Tunel染色發現TC組大鼠出現不同程度的細胞凋亡,染毒后前7 d凋亡指數逐步攀升,7 d達高峰期,TC組與CO組、AC組相同時間點比較差異具有統計學意義,CO組大鼠細胞凋亡也為類似走勢但TC組凋亡指數均低于CO組,且高于AC組。免疫組化結果顯示染毒后大鼠海馬區NQO1的表達從1 d開始逐漸增加,3 d達峰點,且TC組與CO組、AC組每個相同時間點比較差異均具有統計學意義,TC組大鼠海馬CA1區NQO1的表達明顯高于CO組、AC組。我們有理由相信在大鼠急性一氧化碳中毒后體內發生了明顯的氧化應激反應,染毒前期(染毒后3 d)通過每日一次不間斷運用Nrf2激動劑tBHQ,NQO1一定程度高表達的同時細胞凋亡指數有所下降,通過穩定某些保護性蛋白活性、減少自由基生成、減少蛋白酶體降解等作用發揮正性防御作用從而阻止大鼠高級神經功能損傷。
而Morris水迷宮實驗發現14 d開始TC、CO組大鼠穿越平臺頻次減少,活動路徑隨機自由散漫,日趨嚴重,提示大鼠發生了進行性的學習記憶能力障礙。而此時TUNEL染色發現7~28 d兩組大鼠均發生明顯細胞凋亡現象,免疫組化結果顯示染毒3 d后NQO1持續高表達、緩慢下降走勢,且TC組與CO組、AC組每個相同時間點比較均明顯增高,差異均具有統計學意義。同時TUNEL染色結果顯示此時段TC組細胞凋亡指數逐漸較CO組升高,直至28 d水迷宮實驗結果顯示TC組高級神經功能損傷重于CO組,提示在染毒后期盡管細胞內NQO1表達增高發揮了足夠保護作用但仍不能阻止其毒性作用持續、細胞凋亡進展直至凋亡高峰出現,腦細胞因凋亡大量丟失致神經功能缺損,TC組中NQO1持續高表達不但不能發揮正性保護作用反而進一步加重細胞凋亡,高級神經活動發生進行性損害。這與Lee等發現細胞在缺血缺氧環境下,NQO1蛋白的持續過表達抑制細胞存活甚至促進細胞凋亡的觀點不謀而合,其機制可能是持續過表達的NQO1激活AMPK通路,而后遏制mTOR信號通路有關[26]。因免疫組化僅對海馬CA1區NQO1表達進行了統計,為增加結果的準確性我們對整個海馬的NQO1表達進行Western Blot法檢測,得出的結果與免疫組化結果一致。TC、CO組凋亡高峰出現時間推后于NQO1蛋白表達峰點,或許與細胞功能變化到細胞器實質發生變化所需時間有關,而其行為學的改變基本與明顯細胞凋亡發生時間平行,進一步說明在DEACMP發病機制中海馬區細胞發生的遲發性凋亡起著舉足輕重的作用。
大鼠急性CO中毒后,對缺氧反應靈敏的海馬區細胞受到氧化應激作用,前期NQO1表達升高通過穩定保護性蛋白活性、減少自由基生成等途徑防御腦細胞損傷,但后期NQO1持續高表達導致其氧化還原產物氫醌生成達一定濃度、堆積達一定時間誘導凋亡途徑的發生,且細胞持續缺氧難以抵抗鈣離子集聚、炎癥介質釋放等的發生,甚至推動細胞遲發性凋亡的發生發展。經Pearson 直線相關分析顯示CO及TC組NQO1表達與凋亡指數趨勢一致,CO組NQO1與細胞凋亡呈正相關(r=0.505,P<0.001),TC組NQO1與細胞凋亡無相關性(r=0.0049,P<0.001),實質上TC組中兩者并不是沒有相關性,而是NQO1升高與細胞發生凋亡之間密切程度不高,即NQO1高表達在染毒后前期發揮抗凋亡作用和后期發揮促凋亡作用更顯著,將兩時段結合分析得出相關性不高的結果。
近年來臨床中仍有大量急性CO中毒所致精神心理行為異常、智能減退、運動障礙、甚至癱瘓癡呆、尿便失禁、生活不能自理的患者,嚴重影響患者及家人的生活質量,DEACMP致病病理生理途徑多樣且繁雜,是臨床工作者持續面臨的挑戰。研究發現Nrf2/ARE通路在其中起著至關重要的作用,其下游抗氧化蛋白NQO1在一定時間內一定程度的發揮著正性積極作用,而隨著NQO1持續高表達后對細胞的消極負性作用逐漸浮出水面。本實驗為進一步研究其致病機制及靶向治療提供了實驗基礎,為臨床減少DEACMP的發生和研究其靶向治療提供一定的基礎和方向。
[參考文獻]
[1]Yueh-Feng S,Chen MH,Peng GS,et al. Generalized chorea due to delayed encephalopathy after acute carbon monoxide intoxication[J]. Annals of Indian Academy of Neurology,2015,18(1):108-110.
[2]Hu HG,Scholten I,Gruss C,et al. The distribution of the DEK protein in mammalian chromatin[J]. Biochemical & Biophysical Research Communications,2007,358(4):1008-1014.
[3]李勇軍,劉 強,趙春梅,等. 改良式腹腔注射CO法建立急性CO中毒遲發腦病大鼠模型[J]. 寧夏醫科大學學報,2012,34(4):334-336.
[4]趙風華,莊 玉,呼格吉樂圖. 絨山羊肥大細胞類胰蛋白酶的免疫組化及圖像分析研究[J]. 畜牧與飼料科學,2012,33(1):8-11.
[5]Smith MT. Benzene,NQO1,and genetic susceptibility to cancer[J]. Proceedings of the National Academy of Sciences of the United States of America,1999,96(14):7624-7626.
[6]Radjendirane V,Joseph P,Lee YH,et al. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity[J]. Journal of Biological Chemistry,1998,273(13):7382-7389.
[7]Ma Y,Kong J,Yan G,et al. NQO1 overexpression is associated with poor prognosis in squamous cell carcinoma of the uterine cervix[J]. BMC Cancer,2014,14(1):414.
[8]Yang Y,Yan Z,Wu Q,et al. Clinical implications of high NQO1 expression in breast cancers[J]. Journal of Experimental & Clinical Cancer Research Cr,2014,33(1):14.
[9]Siegel D,Gustafson DL,Dehn DL,et al. NAD(P)H:Quinone Oxidoreductase 1:Role as a Superoxide Scavenger[J]. Molecular Pharmacology,2004,65(5):1238-1247.
[10]Shrader WD,Akiko A,Adam B,et al. Towards a modern definition of vitamin E-evidence for a quinone hypothesis[J]. Bioorganic & Medicinal Chemistry Letters,2012,22(1):391-395.
[11]Shih AY,Li P,Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo[J]. Journal of Neuroscience the Official Journal of the Society for Neuroscience,2005,25(44):10321.
[12]Wang YX,Santa-Cruz K,De Carli C,et al. NAD(P)H:quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Alzheimer’s disease [J]. Neurobiol Aging,2000,21:525-531.
[13]Stringer JL,Gaikwad A,Gonzales BN,et al. Presence and induction of the enzyme NAD(P)H:quinone oxidoreductase 1 in the central nervous system. [J]. Journal of Comparative Neurology,2004,471(3):289-297.
[14]Munday R,Smith BL,Munday CM. Effect of inducers of DT-diaphorase on the toxicity of 2-methyl- and 2-hydroxy-1,4-naphthoquinone to rats[J]. Chemico-Biological Interactions,1999,123(3):219-237.
[15]Mcguinness SM,Johansson R,Lundstrom J,et al. Induction of apoptosis by remoxipride metabolites in HL60 and CD34+/CD19- human bone marrow progenitor cells:potential relevance to remoxipride-induced aplastic anemia[J]. Chemico-Biological Interactions,1999,121(3):253-265.
[16]Chevallet M,Wagner E,Luche S,et al. Regeneration of peroxiredoxins during recovery after oxidative stress:only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. [J]. Journal of Biological Chemistry,2003,278(39):37146.
[17]Kuribayashi K,Mayes PA,Eldeiry WS. What are caspases 3 and 7 doing upstream of the mitochondria[J]. Cancer Biology & Therapy,2006,5(7):763-765.
[18]Zhang F,Lau SS,Monks TJ. A Dual Role for Poly(ADP-Ribose) Polymerase-1 During Caspase-Dependent Apoptosis[J]. Toxicological Sciences,2012,128(1):103-114.
[19]Brunet A. Bonni A. Zigmend MJ,et al. Akt promotes cell survival by phospherylating and inhihiting a frokhead transcription factor [J]. Cell,1999,96:857-868.
[20]Zhu J,Wang H,Yang S,et al. Comparison of toxicity of benzene metabolite hydroquinone in hematopoietie stem cells derived from murine embryonic yolk sae and adult bone marrow[J]. PLoS One,2013,8:e71153.
[21]Ying MD,Tu CX,Ying HZ,et al. MSFTZ,a flavanone derivative,induces human hepatoma cell apoptosis via a reactive oxygen species-and caspase-dependent mitochondrial pathway[J]. Journal of Pharmacology and Experimental Therapeutics,2008,325:758-765.
[22]Nogueira V,Park Y,Chen CC,et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis[J]. Cancer Cell,2008,14(6):458-470.
[23]Keum YS,Han YH,Liew C,et al. Induction of heme oxygenase-1(HO-1)and NAD [P]H:quinone oxidoreductase 1 (NQO1)by a phenolic antioxidant,butylated hydroxyanisole (BHA)and its metabolite,tert -butylhydroquinone (tBHQ)in primary -cultured human and rat hepatocytes[J]. Pharm Res,2006,23:2586-2594.
[24]武小媛,曲麗艷,全 康,等. tBHQ和Sulforaphane對Caco2細胞Nrf2-ARE信號通路的影響[J]. 浙江大學學報(醫學版),2010,39(1):17-23.
[25]Sun Q,Cai J,Zhou J,et al. Hydrogen-rich saline reduces delayed neurologic sequelae in experimental carbon monoxide toxicity[J]. Critical Care Medicine,2011,39(4):765-769.
[26]Lee H,Oh ET,Choi BH,et al. NQO1-induced activation of AMPK contributes to cancer cell death by oxygen-glucose deprivation[J]. Sci Rep,2015,5:7769.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
創新作文(小學版)(2019年10期)2019-09-25 08:12:28
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
西南軍醫(2016年6期)2016-01-23 02:21:19
作文與考試·小學高年級版(2015年17期)2015-05-30 10:48:04
