(1S,4R)- 4,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷-1,4-二醇的一步催化合成及其除草活性
2018-06-20 12:58:48謝志鵬黃道戰黃燕培黃燕妮
生物質化學工程 2018年3期
關鍵詞:催化劑
謝志鵬, 黃道戰, 黃燕培, 黃燕妮
(廣西民族大學 化學與化工學院, 廣西 南寧 530008)
5,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷- 4-醇(1)是含有6-氧雜二環[3.2.1]辛烷骨架的雙環單萜含氧化合物,具有良好的除草活性[1-2]。然而,由于其合成原料α,α,3-三甲基-3-環己烯-1-甲醇來源少、制備成本高,化合物1在產業化及應用推廣上嚴重受限。基于化合物1的分子結構特征、合成原理以及農業生產對植物源除草劑的需求,人們以資源豐富、價廉易得的單萜烯烴及其含氧衍生物為原料,合成了具有6-氧雜二環[3.2.1]辛烷骨架的系列雙環單萜含氧化合物并對化合物的除草活性進行了研究[3-7]。但是,目前此類雙環單萜含氧化合物在品種和數量上還較少,其生物活性方面的研究也不夠深入。(1S,4R)- 4,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷-1,4-二醇(2)含有6-氧雜二環[3.2.1]辛烷骨架和2個羥基,在分子結構上與化合物1十分相似,目前國內外尚無對其除草活性的研究報道。另外,化合物2的合成方法[8]仍是采用兩步或多步化學合成工藝,即以由α-蒎烯經酸催化異構制成的異松油烯為原料或直接以天然異松油烯為原料,經過氧酸環氧化、酸催化環氧和開環重排反應制備,制備過程存在工藝復雜、操作安全性差等不足。本課題組前期研究采用具有氧化-酸雙功能催化作用的過氧磷鉬鎢酸十六烷基吡啶鹽為催化劑,在H2O2存在下,催化α-蒎烯一步轉化合成得到了(3R,4R)- 4,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷-3,4-二醇[5],在此基礎上,本研究在過氧磷鉬鎢酸十六烷基吡啶鹽催化作用下,采用異松油烯與H2O2反應合成得到化合物2,并對化合物2的結構進行了表征,進一步分析了其對常見雜草黑麥草的除草活性。
1 實 驗
1.1材料與儀器
異松油烯,購于江西環球天然香料有限公司,純度為96.1 %。催化劑過氧磷鉬鎢酸十六烷基吡啶鹽[π-C5H5N(CH2)15CH3]3PMo2W2O24(簡稱HPP)按文獻[9~10]方法制備。質量分數30 %的H2O2、氯仿、乙酸乙酯等均為市售分析純。黑麥草,購自美國百綠集團。
GC-2014C型氣相色譜儀,島津(蘇州)儀器有限公司;Magna IR 550型紅外光譜(IR)儀,美國Nicolet公司;Advance 600型低溫超導核磁共振波譜(NMR)儀,瑞士Bruker公司;Smart APEX Ⅱ型單晶X射線衍射(XRD)儀,德國Bruker公司;LRH-250-GSI人工氣候箱,廣東韶關市泰宏醫療器械有限公司。
1.2樣品合成
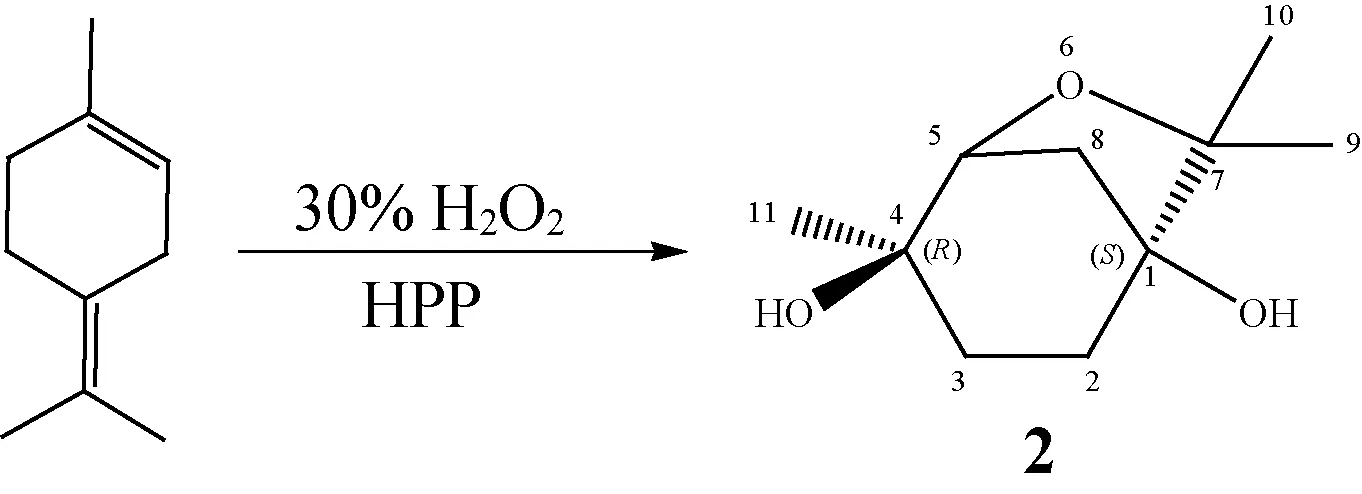
向25 mL恒溫水浴夾套玻璃反應器中依次加入異松油烯(0.54 g,4 mmol)、催化劑HPP、H2O2和溶劑(0.8 mL),在一定溫度下磁力攪拌反應一段時間。反應結束后,待反應混合物冷卻至室溫,加入一定質量的內標物萘和溶劑無水乙醇,充分溶解混合,靜置使催化劑沉淀析出,取澄清液直接進行氣相色譜定量分析,采用內標法計算異松油烯的轉化率和目標產物的產率。化合物2的合成反應式如右所示。
1.3樣品結構表征
1.3.1XRD單晶結構表征 將1.2節中反應混合物冷卻后蒸發脫水濃縮,然后用乙酸乙酯溶解并過濾,濾液冷卻結晶得到粗產品,再將粗產品重結晶6次,然后用乙酸乙酯將所得晶體溶解配制成稀溶液,采用溶劑揮發法在室溫下培養單晶,選取一定尺寸(0.20 mm×0.20 mm×0.05 mm)的單晶樣品,進行XRD分析。采用石墨單色化Mo Kα射線(λ=0.071 073 nm),在溫度為296.15 K條件下采集數據,采用多重掃描法對采集的數據進行吸收校正,直接法解出結構,全矩陣最小二乘法對非氫原子坐標及各向異性熱參數進行精修[11],其中氫原子均為理論加氫。
1.3.2IR表征 將1.3.1節中的粗產品經4次重結晶后,取少量采用KBr壓片法,獲取紅外吸收光譜。
1.3.3NMR表征 將1.3.1節中的粗產品經4次重結晶后,取少量以氘代氯仿為溶劑,在核磁共振波譜儀中測定其1H NMR和13C NMR。
1.4除草活性測試
采用平皿培養法[12-13],以黑麥草為研究對象,初步評價化合物2的除草活性。將適量黑麥草種子在無菌水中浸泡18 h后,置于人工氣候箱中,在28 ℃下催芽萌發露白。在直徑為9 cm的培養皿中鋪放一張直徑為8.5 cm的消毒紗布,加入10 mL一定濃度的供試化合物溶液和10粒剛萌發露白的黑麥草種子,每個濃度溶液做3組平行實驗,以等體積的無菌水作為空白對照液,在人工氣候箱中28 ℃下遮光培養72 h,測量、統計胚根和胚芽的平均長度,按公式(1)和(2)分別計算根長或芽長抑制率,評價化合物2的除草活性。

(1)

(2)
2 結果與分析
2.1合成反應條件的討論
2.1.1溶劑種類 在異松油烯用量4 mmol、反應物料比(H2O2與異松油烯物質的量比,下同)值為3、催化劑用量5.0 %(以異松油烯質量計,下同)、反應溫度30 ℃、溶劑用量0.8 mL、反應時間3 h的條件下,考察溶劑種類對反應轉化率和產率的影響,結果列于表1。由表1可知,溶劑種類明顯影響反應轉化率和產率,以極性的乙醇、中極性的乙酸乙酯和丙酮以及非極性的正己烷為溶劑時,反應轉化率和產率都較低,而以弱極性的氯仿和1,2-二氯乙烷為溶劑,轉化率和產率均高于其他溶劑,其中轉化率均高達100 %。究其原因可能與催化劑在不同溶劑中的溶解情況有關,催化劑HPP是一種相轉移催化劑[10],在分子結構上是由十六烷基吡啶陽離子和過氧磷鉬鎢酸陰離子兩部分按3∶1的物質的量比組成,即存在3條疏水性的十六烷基長鏈將親水性的陰離子“包埋”的情況,有機溶劑種類明顯影響“包埋”情況,進而影響催化劑的溶解性能及催化性能。催化劑HPP在極性、中極性和非極性溶劑中溶解性差,催化活性不高,轉化率較低,而在弱極性鹵代烴溶劑中溶解性較好,并表現出良好的相轉移催化作用。親水性雜多陰離子能夠與水相中的雙氧水反應生成“活性氧物種”,然后作為反離子與親油性的十六烷基吡啶陽離子結合,被帶到有機相,與底物接觸反應。鹵代烴溶劑中,與氯仿相比,1,2-二氯乙烷易燃、毒性較大,因此,選擇氯仿作為反應溶劑。
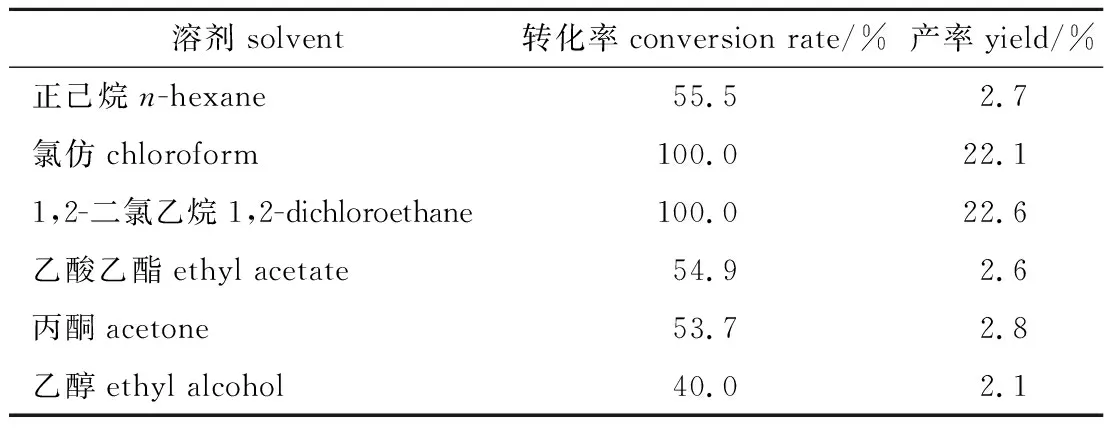
表1 溶劑種類對轉化率和產率的影響
2.1.2溫度 以氯仿為溶劑,其他反應條件同2.1.1節,考察溫度對反應轉化率和產率的影響,結果如圖1(a)所示。由圖1(a)可知,溫度對反應轉化率和產率有一定的影響,溫度為20 ℃時,轉化率和產率很低,分別為66.0 %和3.4 %;溫度高于25 ℃,隨著溫度升高,轉化率和產率先快速提高而后趨于平穩或緩慢降低,其中轉化率在30 ℃時就已達到100 %而后保持不變,而產率先由5.8 %(25 ℃)提高到25.7 %(35 ℃),然后緩慢降低。這是由于異松油烯具有2個活潑的碳碳雙鍵,常溫下容易發生氧化反應,而氧化劑過氧化氫屬于過氧化物,較低溫度下相對穩定,需達到一定溫度才能發生自分解,釋放“活性氧”參與反應,但當溫度高于35 ℃時,過氧化氫自分解反應加劇,環氧和開環水解反應加速,副產物增多,化合物2的產率下降。因此,反應溫度選取35 ℃為宜。
2.1.3催化劑用量 反應溫度35 ℃,其他反應條件同2.1.2節,考察催化劑用量對反應轉化率和產率的影響,結果如圖1(b)所示。由圖1(b)可知,催化劑用量對轉化率和產率有較大的影響。不添加催化劑,轉化率和產率較低。隨著催化劑用量由0增加至9.19 %時,轉化率呈先增加后不變的趨勢,化合物2的產率大致呈先增加后略微減小的趨勢。催化劑用量為7.35 %時,轉化率為100 %,同時產率達到最大值,為27.1 %。原因可能是隨著催化劑用量的增加,異松油烯發生高效的氧化反應,但用量的增加會使反應體系酸量增加,環氧和開環水解反應加速,副產物增多,故化合物2的產率下降。因此,本實驗的最佳催化劑用量為7.35 %。
2.1.4反應物料比 催化劑用量為7.35 %,其他反應條件同2.1.3節,考察反應物料比對反應轉化率和產率的影響,結果如圖1(c)所示。由圖1(c)可知,反應物料比對反應轉化率和產率有一定的影響,隨著反應物料比值由1增加至5,轉化率由88.3 %很快提高至100 %并保持不變,而產率呈先增后減的變化趨勢。當反應物料比值為3時,轉化率為100.0 %,同時產率達到最大值,為27.1 %。究其原因可能是異松油烯轉化生成目標產物的反應經歷環氧化、酸催化開環重排,催化反應體系需要一定的酸性條件,但酸性太強或酸量過多對轉化反應不利。實驗發現,隨著反應物料比值增加,即自身具有一定酸性的H2O2用量增多,反應體系酸量增加,環氧和開環水解反應加速,生成了較多的雙羥基化、脫水等副產物,目標產物產率降低。故反應物料比值選擇3為宜。
2.1.5反應時間 反應物料比值為3,其他反應條件同2.1.4節,考察反應時間對轉化率和產率的影響,結果如圖1(d)所示。由圖1(d)可知,反應時間對轉化率的影響不明顯,而對產率的影響較明顯;反應0.5 h,轉化率就達到99.6 %,而產率才達到13.1 %,隨著反應時間延長至6 h,產率先緩慢增加至29 %而后略微降低。究其原因,異松油烯具有2個活潑的碳碳雙鍵,容易發生氧化反應,生成環氧化物中間體,中間體再經開環、水解、重排等系列反應,最終生成目標化合物2,適當延長反應時間,有利于產率的提高。因此,反應時間選擇6 h。
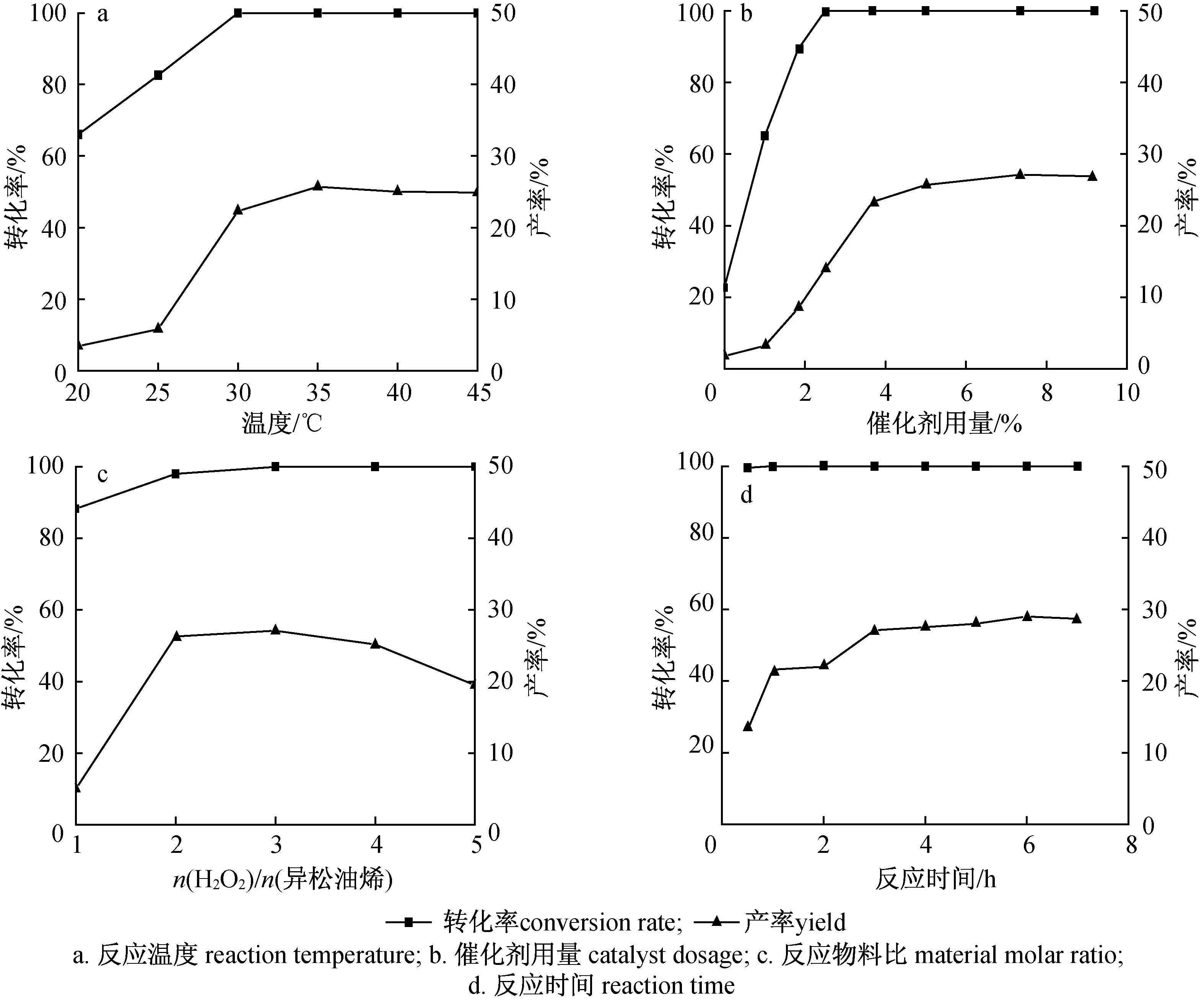
圖1 反應條件對轉化率和產率的影響Fig. 1 Effect of reaction conditions on the conversion rate and yield
研究發現較佳的反應條件是異松油烯用量4 mmol,催化劑HPP用量7.35 %,溶劑氯仿用量0.8 mL,反應物料比值3,于35 ℃反應6 h,此條件下異松油烯的轉化率高達100 %,目標產物的產率達到29 %,產率不高,催化劑及催化反應體系有待進一步改進,以提高目標產物的產率。
按上述反應物料比擴大125倍,催化劑用量5.0%,其他條件相同進行反應。反應結束后,冷卻至室溫,靜置、分液,分出水相,減壓蒸發脫水濃縮,得淺黃色油狀黏稠物;該黏稠物用適量乙酸乙酯加熱溶解,過濾除去不溶物;濾液冷卻結晶,減壓過濾收集得到粗產品;將粗產品重結晶4次,真空干燥得到白色粉末晶體產品,得率為8.47 %,純度(氣相色譜峰面積歸一化法)達到98.8 %。
2.2化合物2的結構表征
2.2.1紅外光譜表征 化合物2紅外光譜圖如圖2所示,其主要特征峰及歸屬為:3367 cm-1為O—H的伸縮振動吸收峰;2965和2928 cm-1為甲基和亞甲基的C—H不對稱伸縮振動吸收峰;1135 cm-1為C—O的伸縮振動吸收峰;1063 cm-1為C—O—C的伸縮振動吸收峰。IR表明,目標產物中含有羥基和醚鍵等官能團。
2.2.2核磁共振表征 化合物2的NMR譜見圖3。1H NMR(CDCl3為溶劑,600 MHz)δ:3.72~3.75(1H,t,H-5),2.23(1H,s,OH-1),2.22(1H,s,OH- 4),1.90~1.91(2H,d,H-8),1.76~1.77(2H,t,H-3),1.58~1.60(2H,t,H-2),1.24(3H,s,H-11),1.24(3H,s,H-10),1.20(3H,s,H-9);13C NMR譜(CDCl3為溶劑,150 MHz)δ:21.25(C-9),26.79(C-10),27.96(C-11),32.56(C-2),34.19(C-3),37.05(C-8),71.64(C- 4),77.50(C-5),80.81(C-1),81.39(C-7)。由NMR分析可知,目標產物有18個H和10個C,與理論值相符。
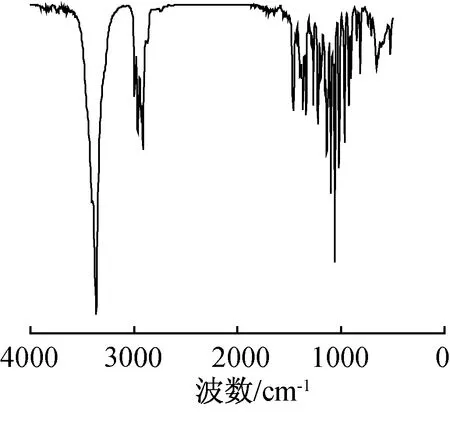
圖2 化合物2的IR譜圖Fig. 2 IR spectrum of compound 2
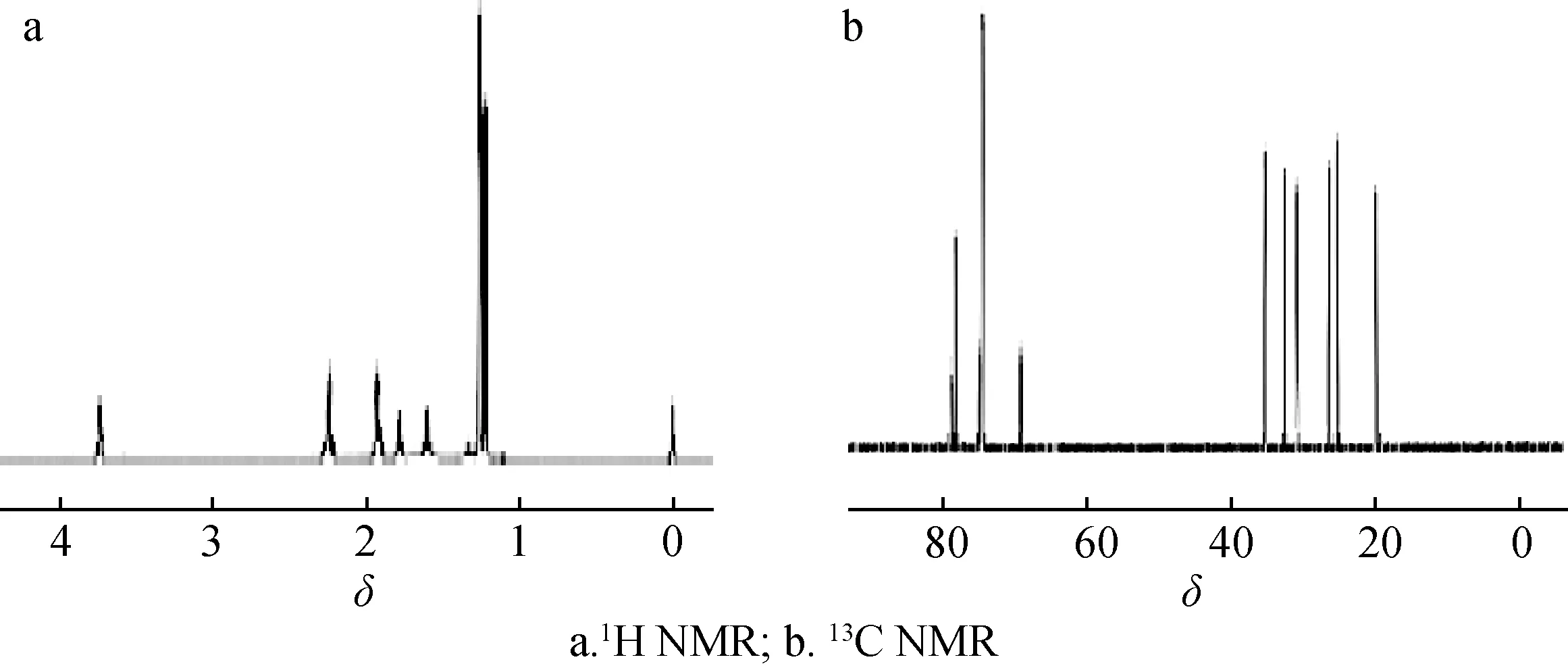
圖3 化合物2的核磁共振譜圖Fig. 3 NMR spectra of compound 2
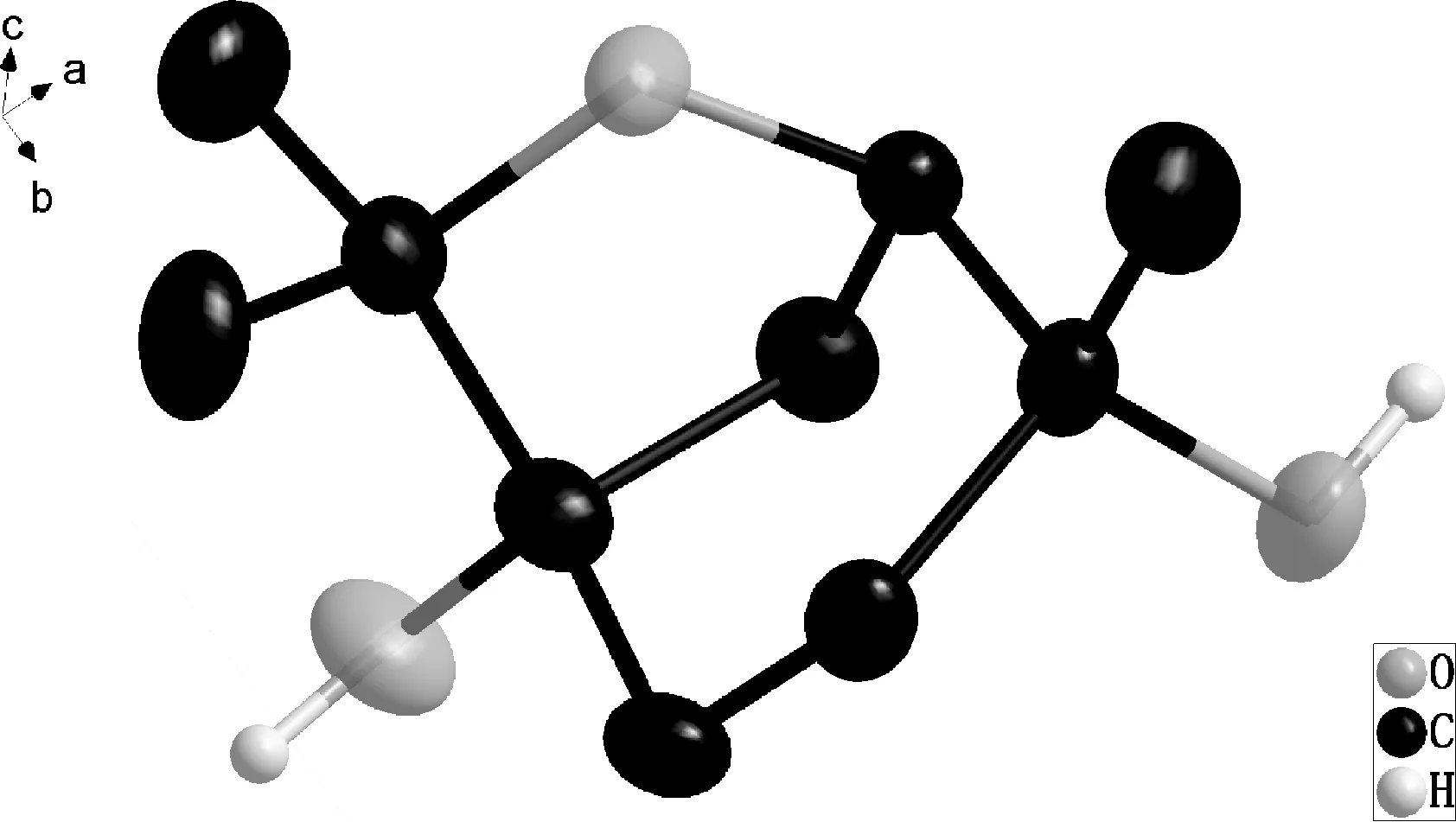
圖4 化合物2的分子晶體結構圖Fig. 4 Molecular crystal structure of compound 2
2.2.3XRD表征 對化合物2進行單晶X射線衍射分析,其分子晶體結構如圖4所示,顯示該化合物是一種含有一個五元含氧雜環和一個環己烷的單萜6-氧雜雙環[3.2.1]辛烷類衍生物,擁有3個手性碳原子,其中手性碳原子C1和C4均連接有一個羥基,分別為S構型和R構型。據此證實了該化合物分子的立體結構,按照系統命名法,其名稱為(1S,4R)- 4,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷-1,4-二醇,這也與多步合成方法[8]的結果相符。
2.3除草活性
采用2.1節中較佳條件下重結晶提純后得到的化合物2對黑麥草進行除草活性實驗,結果如表2所示。由表2可知,化合物2能明顯抑制黑麥草的根和芽的生長,其抑制作用隨著濃度的升高而增強。濃度低于80 mmol/L,芽長的抑制率高于根長的抑制率,即化合物2對芽生長的抑制作用大于對根生長的抑制作用;而濃度高于80 mmol/L,芽長的抑制率稍低于根長的抑制率,即化合物2對芽生長的抑制作用小于對根生長的抑制作用;可能是在低濃度時芽對化合物2的吸收強于根,所以化合物2對芽長的抑制率高于根,而在高濃度時,情況正好相反。在濃度為120 mmol/L時,化合物2對黑麥草的根長和芽長的抑制率分別高達91.8 %和80.7 %,表現出較高的除草活性。在相同濃度下,與已報道的結構類似的1,8-桉樹腦[14]相比,化合物2對黑麥草的抑制作用更好。
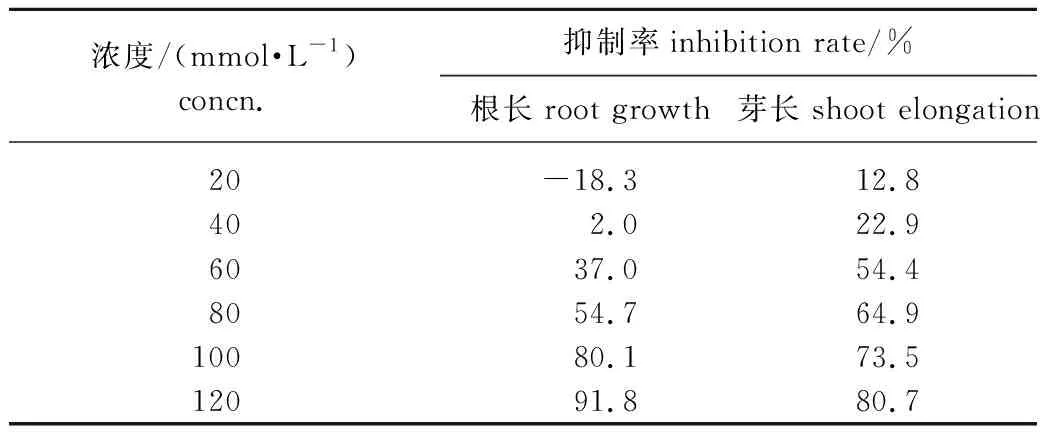
表2 化合物2對黑麥草的除草活性Table 2 Herbicidal activity of compound 2 against ryegrass
3 結 論
3.1以過氧磷鉬鎢酸十六烷基吡啶鹽(HPP)為氧化-酸雙功能相轉移催化劑,在30 %(質量分數)H2O2存在下,催化異松油烯一步合成(1S,4R)- 4,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷-1,4-二醇。溶劑種類、反應溫度、反應物料比、催化劑用量和反應時間對轉化率和產率有較大影響,研究發現較佳的反應條件為異松油烯用量4 mmol、催化劑HPP用量7.35 %,溶劑氯仿用量0.8 mL、反應物料比值3、35 ℃反應6 h,此條件下異松油烯的轉化率高達100 %,目標產物的產率達到29 %。
3.2紅外光譜、1H核磁共振譜、13C核磁共振譜及單晶X射線衍射的分析結果表明成功合成了化合物2。
3.3化合物2能抑制黑麥草的根和芽的生長,其抑制作用隨著濃度的升高而增強。在120 mmol/L的濃度下,化合物2對黑麥草根和芽的生長的抑制率分別達到91.8 %和80.7 %,表現較高的除草活性。
參考文獻:
[1]POWELL J E. 6-Oxabicyclo[3.2.1]octane derivatives and compositions and methods for controlling plant growth:US 4486219[P]. 1984-12- 04.
[2]POWELL J E. Trimethyl 6-oxabicyclo[3.2.1]octan- 4-ols and 4-ones:US 4536586[P]. 1985- 08-20.
[3]POPOVA L A,BIBA V I,PRISHCHENPENKO V M,et al. Synthesis of monoterpene oxabicyclodiols starting fromα-pinene[J]. Journal of General Chemistry of the USSR in English Translation,1992,62(72):1346-1350.
[4]COSTA V V,ROCHA K A,SOUSA L F,et al. Isomerization ofα-pinene oxide over cerium and tin catalysts:Selective synthesis of transcarveol andtrans-sobrerol[J]. Journal of Molecular Catalysis A:Chemical,2011,345(1/2):69-74.
[5]黃道戰,朱守記,藍虹云,等. 一種制備(3R,4R)- 4,7,7-三甲基- 6-氧雜二環[3.2.1]辛烷-3,4-二醇的新方法[J]. 林產化學與工業,2015,35(1):9-15.
[6]黃道戰,朱守記,藍虹云,等. 雙官能單萜氧雜二環二醇乙酸酯類衍生物的合成及其除草活性[J]. 合成化學,2015,23(3):185-190.
[7]ZHAO Z Y,HUANG D Z,LAN H Y,et al. Novel synthesis, crystal structure and herbicidal activity of (3R,4R)- 4,7,7-trimethyl- 6- oxabicyclo[3.2.1]octane-3,4-diol[J]. Chinese Journal of Structure Chemistry,2015,34(12):1819-1824.
[8]SALOMATINA O V,YAROVAYA O I,KORCHAGINA D V,et al. Acid-catalyzed transformations of diepoxy derivatives of terpinolene[J]. Russian Journal of Organic Chemistry,2011,47(10):1479-1486.
[9]黃道戰,農秀李,黃雪梅,等. 過氧磷鉬鎢酸鹽催化一步法合成2-羥基-1,4-桉樹腦[J]. 精細化工,2015,32(10):1185-1189.
[10]ZHAO W,MA B,HUA H,et al. Environmentally friendly and highly efficient alkenes epoxidation system consisting of [π-C5H5N(CH2)11CH3]3PW4O32/H2O2/ethyl acetate/olefin[J]. Catalysis Communications,2008,9(14):2455-2459.
[11]SHELDRICK G M. Shelxl-97, Program for X-ray Crystal Structure Refinement[M]. Germany:University of G?ttingen,1997.
[12]烏婧,王寶雷,李永紅,等. N-芐氧/烷氧苯基- 4,6-二取代嘧啶胺類化合物的合成、晶體結構及除草活性[J]. 高等學校化學學報,2008,29(8):1583-1587.
[13]黃道戰,王小淑,朱守記,等. 對孟烷-1,2,4-三醇的合成及對水稻稗草的除草活性研究[J]. 生物質化學工程,2013,47(5):40-44.
[14]BARTON A F M,CLARKE B R,DELL B,et al. Post-emergent herbicidal activity of cineole derivatives[J]. Journal of Pest Science,2014,87(3):531-541.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
