三維自支撐電極在析氫反應中的研究進展
2018-05-14 02:28:19譚勇文
中國材料進展 2018年4期
關鍵詞:催化劑
趙 揚,彭 鳴,譚勇文
(湖南大學材料科學與工程學院,湖南 長沙 410082)
1 前 言
隨著石油、煤、天然氣等傳統化石能源的急劇消耗,其燃燒所帶來的環境污染、溫室氣體大量排放等問題日益凸顯。因此,開發環境友好、清潔高效的可替代新能源勢在必行。氫氣作為能量密度高、燃燒產物無污染、可再生的綠色能源,在應對能源和環境問題時被寄予厚望,受到廣泛關注[1-3]。傳統的工業制氫方法主要是甲烷蒸汽重整和煤的氣化,這兩種方法制得的氫氣占到氫氣總產量的90%以上,但其仍然需要大量的化石能源作為原料。而電解水制氫具有裝置簡單、產物純凈、能量轉化率高等特點。特別是隨著風能、太陽能、潮汐能等可再生能源發電技術的進步,電解制氫的便捷性將大大提高、成本有望進一步降低,有望成為未來能源發展的重要方向[4-8]。
在電解水制氫(HER)的反應中,電極的性質包括電極的表面積、導電性、催化活性和穩定性等,都對析氫性能影響顯著[9]。常見的電催化材料大多為粉體,需要在溶劑中分散后,利用有機粘接劑(如Nafion和PVDF)將其固定在玻碳電極上。但工作電極面積受限于玻碳電極的面積,催化劑的負載量有限;同時,粘結劑會降低材料的導電性,另一方面會掩蓋催化劑的部分活性位點,降低催化性能;長時間電解過程中,活性催化材料易脫落,影響電極的穩定性。高性能析氫電極的一個最新發展趨勢是,引入多孔基底以增大活性表面積、利用原位催化劑制備獲得良好的界面接觸,以減少粘接劑和導電劑的使用,獲得了性能優異的“三維自支撐電極”。相比于傳統活性材料修飾的玻碳電極,三維無粘接劑型自支撐電極具有更大的活性比表面積,能有效地增加活性物質的負載量,擁有更多的表面活性催化位點,有利于電子傳輸和質子轉移,從而展示出優異的催化性能[10-12]。同時,三維自支撐材料具有制備工藝簡單、性能穩定等優點,具有良好的應用前景。
因此,本文概述了近年來三維自支撐電極在析氫反應中的研究進展。內容涵蓋了電極材料的分類、電極材料催化活性的評價方法、電極材料的制備及其電解析氫性能等,總結了三維自支撐電解水析氫催化劑面臨的挑戰,展望了未來的發展方向。
2 電解水析氫反應概述
2.1 電解析氫材料
析氫反應是電化學催化反應中最基本的反應之一。近年來,發展出了各類催化電極材料,根據不同的空間維度和結構形貌,電極材料可分為:零維的納米團簇、納米原子、量子點等;一維的納米線、納米管、納米棒等;二維的納米帶、納米片、薄膜等;三維的納米陣列、自支撐多孔電極材料等。各類電極材料得到研究者們廣泛的研究和探索,其性能不斷提高,有望取代貴金屬鉑,實現低成本的電解析氫應用[13, 14]。隨著材料制備技術的發展以及電解水工業的實際需求,基于非貴金屬材料的三維自支撐電極是當前研究的一個熱點。不同維度電極材料的性能對比見表1。

表1 不同維度的析氫電催化劑的催化活性
Notes: QDs: quantum dots, GQDs: graphene quantum dots, NGCLs: N-doped graphitic carbon layers, RGO: reduction of graphene oxide, NWs: nanowires, HNW: hollow nanowires, CC: carbon cloth, CFP: carbon fiber paper.
2.2 電極催化活性的評價指標
析氫反應的機理得到較多研究,無論是Volmer-Heyrovsky機理還是Volmer-Tafel機理,在不同的催化體系不斷得以印證和完善,為新材料和新結構的設計提供了重要依據[31, 32]。一般通常采用總電極活性、塔菲爾曲線、電化學阻抗、穩定性、轉換頻率和法拉第效率等參數來評估電解析氫電極體系中功能電極的催化性能[33-35]。
(1)總電極活性。通常以一定電壓下的穩定電流密度或一定電流密度下的過電勢作為比較的標準。其中,電流密度為10 mA/cm2時的過電勢最為常見。
(2)塔菲爾曲線。記錄穩態時,析氫過電勢與電流密度的關系。以過電勢(η)對電流密度(j)的對數進行線性擬合,根據塔菲爾方程(η=a+blogj)擬合得到斜率,b值通常被認為與反應機理有關。當過電勢趨近于零時,計算得出j0,即為交換電流密度,反映催化材料本征的催化活性。塔菲爾斜率越小,交換電流密度j0越大,電極催化性能越好。
(3)電化學阻抗譜。電化學阻抗譜是一種探究HER動力學和電極/電解液界面反應的電化學分析手段,高頻區對應電荷轉移阻抗,較低的電荷轉移電阻值意味著快速的反應速率。
(4)穩定性。在一定酸堿條件下電解析氫反應的持續性,是反映電極結構和性能穩定的重要指標。可以通過觀測多次重復測試CV或LSV曲線的重合性,或者持續監測一定電壓(電流)下的電流(電壓)來評估電極材料的穩定性。
(5)轉換頻率。轉換頻率是指單位時間內在單位催化活性位點上的反應物分子轉化數,該值越大,表明材料的催化產氫速率越快。
(6)法拉第效率。法拉第電流效率是指析氫過程中的實際產氫量與理論產氫量的比值,用于衡量電流利用率的高低,同時也能衡量材料的穩定性。理想的電解水催化劑的法拉第效率為100%。
3 三維自支撐電極材料的研究進展
3.1 金屬為襯底的三維催化電極
金屬為襯底的三維催化電極具有高的比表面積、高導電性和廉價易得等特點,本小節主要總結了以泡沫鎳(Ni foam,NF)、泡沫銅(Cu foam,CF)、不銹鋼片、鈦網等為代表的以金屬為襯底的三維電極在析氫領域中的應用。
3.1.1 泡沫鎳襯底
泡沫鎳是一種具有優良性能的多孔材料,其三維網狀結構使其具有高達1098~9146 cm2/cm3的比表面積,且其在堿性條件下穩定性好,因此被廣泛用作三維析氫電極的基底材料[36-38]。
酸性條件對鎳基底有強腐蝕性,會致使催化劑穩定性嚴重下降。因此研究者們往往會對泡沫鎳進行表面處理,防止其被腐蝕,以期得到性能良好的電極材料。早在2013年,Chang等[39]就研究了石墨烯包覆泡沫鎳在酸性條件下的析氫性能。研究發現石墨烯包覆不僅提高了活性物質的負載量,同時還提高了電極的析氫活性和穩定性。作者采用CVD法在泡沫鎳上沉積了一層無定型硫化物(MoSx,x>2)后,在酸性電解質條件下,該材料的析氫過電勢為0.2 V,Tafel斜率為42.8 mV/dec,產氫速率達到13.47 mmol·g-1·cm-2·h-1。Ren課題組將泡沫鎳直接硒化改性為三維多孔硒化鎳泡沫材料,在酸性條件析氫方面做了大量研究工作[40-42]。Zhou等[43]采用硒化改性后的鎳泡沫,以(NH4)2MoS4為硫源和鉬源,在500 ℃管式爐中共沉積硒化得到MoSSe三元化合物(MoSSe/NiSe2),10 mA/cm2電流密度下的過電勢為69 mV,Tafel斜率為42.1 mV/dec,交換電流密度為299.4 μA/cm2。最近,Zhou等[44]采用醋酸對泡沫鎳進行表面預處理后,直接以泡沫鎳作為鎳源硒化,得到的材料的性能(Tafel斜率為42.6 mV/dec)可與貴金屬鉑相媲美。更為重要的是,此方法為制備大批量、廉價易得的產氫電極材料提供了思路。
相比起酸性條件,金屬基底在堿性和中性條件下比較穩定。但由于溶液電阻較大,反應能壘較高,導致動力學反應較慢,影響了總的反應速率。為了提高在堿性、中性條件下的析氫性能,降低反應過電勢等,研究者們開展了大量的研究工作[45-48],并取得了極大的突破。2014年,Geng等[49]在石墨烯處理后的泡沫鎳上垂直生長MoS2薄片陣列,在堿性條件下,其起始過電位為25 mV,Tafel斜率為98 mV/dec。Yan等[50]采用溶劑熱法在泡沫鎳上生長Co3O4納米片,之后還原得到Co/Co3O4核殼結構,其起始過電位為30 mV,10 mA/cm2時的過電位為90 mV,Tafel斜率為44 mV/dec。Feng課題組[29]在堿性條件析氫領域取得重大突破,研究人員對泡沫鎳上的NiMoO4長方體前驅體進行退火處理,通過控制Ni原子向外擴散的程度,在泡沫鎳表面得到長方體狀納米MoO2支撐的MoNi4電催化劑(MoNi4/MoO2/NF),具體合成路線如圖1a所示。理論和實驗結果都表明,MoNi4表面發生快速析氫過程,在1.0 M KOH電解液中,起始過電位為0,在10 mA/cm2條件下過電位僅為15 mV(圖1b),Tafel斜率為30 mV/dec (圖1c),催化性能優于同條件下的Pt催化劑,并超過目前所有非Pt催化劑在堿性條件下的催化性能。這種制備方法簡便、成本廉價的催化劑,為堿性電解槽析氫帶來了新的希望。類似的工作(MoNi4/MoO3-x/Ni)和相近的實驗結果(η10=17 mV,Tafel斜率為36 mV/dec)也被Chen等[51]隨后報道。
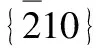
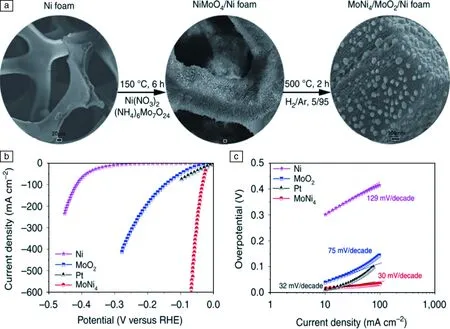
圖1 MoNi4/MoO2/Ni合成示意圖(a),電化學極化曲線(b),Tafel斜率圖(c)[29]Fig.1 Schematic of preparation process of MoNi4/MoO2/Ni (a), polarization curves (b), corresponding Tafel slopes (c) [29]
3.1.2 其它金屬材料襯底
除了以泡沫金屬鎳為襯底的三維自支撐材料外,其它導電材料,比如泡沫銅[61-62]、鈦網/箔[63-65]、鉬網[66]、不銹鋼網[67]、導電玻璃[68-71]等也常常被用作合成三維材料的襯底材料。孫旭平課題組在Ti網[72]、Ti箔[73]、Ti片[74]、Ti板[75]上做了大量研究工作。最近其課題組的Lu等[76]利用硼氫化鈉(NaHB4)原位處理生長在Ti網上的CoO納米線陣列,得到Co-B納米顆粒修飾的三維自支撐電極(Co-B/CoO/Ti)。在堿性條件下,電流密度達到50 mA/cm2時,析氫過電位僅為102 mV,Tafel斜率為78 mV/dec(此實驗測得Pt/C電極Tafel斜率為68 mV/dec),并且具有長時間的穩定性,電解20 h后電流密度僅衰減17%。將其作為兩電極進行堿性水解,電流密度為50 mA/cm2的電壓為1.67 V,電解20 h后電流密度衰減19%。該工作為堿性條件下全解水提供了新思路,金屬硼化物的引入大大提高了其催化性能。泡沫銅用作三維襯底材料,既可以作為襯底,又可以作為銅源。Liu等[77]直接采用化學方法沉積無定型硫化鈷薄膜,在沉積過程中,泡沫銅作為銅源,銅原子通過自擴散進入到薄膜中去,得到亞納米銅簇修飾的硫化鈷三維電極(Cu/CoSx/CF)。在堿性條件下,電解水析氫半反應電流密度為10 mA/cm2和100 mA/cm2的過電勢分別為134 mV和267 mV。將其作為陰極和陽極進行全解水反應,電流密度達到10 mA/cm2和100 mA/cm2時的電壓分別為1.5 V和1.8 V,優于Pt/C-IrO2電極的1.55 V和1.8 V,作者首次證實了銅簇和復合薄膜結合能促進電子轉移,提高催化性能。Yu等[78]從催化劑的微觀形貌設計入手,將銅納米線和鎳鐵雙金屬層狀氫氧化物(NiFe LDH)復合,在泡沫銅基底上巧妙地制備了兩者的三維核殼納米結構(圖2a)。Cu納米線優異的導電性有利于電子從Cu納米線向NiFe LDH納米片轉移。另外,NiFe LDH具有獨特的層狀結構,層與層之間的間隙為氣體分子的快速釋放提供了開放的通道。在1.0 M的KOH溶液中,該電極產生10 mA/cm2電流密度對應的析氫過電位為116 mV,將該電極應用于全分解水反應,在1.54 V和1.69 V的電壓下就能分別產生10 mA/cm2和100 mA/cm2的電流密度(圖2b),優于已報道的大多數非貴金屬催化劑。

圖2 三維Cu@NiFe LDH核殼電極示意圖(a),全分解水極化曲線(b)[78]Fig.2 Schematic of three-dimensional Cu@NiFe LDH core-shell electrode (a), polarization curves for overall water splitting (b) [78]
以金屬作為三維襯底材料制備催化電極的方法具有簡單、耗能低、無污染等優勢,而且適合大尺寸樣品的制備,具有良好的應用前景。但是三維金屬電極在酸性條件下極易被腐蝕,限制了其應用范圍;且以金屬電極為金屬源也將大大降低三維電極的機械強度,從大規模商業化應用的角度來看,如何提高三維電極材料的機械強度和穩定性將是未來發展方向之一。
3.2 碳材料為襯底的三維催化電極
與金屬為基底的三維電催化劑相比,三維碳纖維材料具有廉價易得、柔韌性好、耐酸耐堿等優點。本小節重點綜述了以碳纖維材料(包括碳布(Carbon Cloth,CC),碳纖維紙(Carbon Fiber Paper,CFP)和其它碳材料為基底的三維自支撐催化劑在析氫領域的應用。
3.2.1 碳布襯底
碳布是由碳纖維絲機織而成,具有高導電性和柔韌性。研究者直接在碳布基底上原位生長出各種納米纖維線/棒/桿/球/片狀等陣列材料[79-83],得到三維立體結構,展示了良好的催化析氫效果。但經過多年發展,人們已經不滿足于簡單的水熱生長、電化學沉積單一的金屬或金屬復合物在碳布基底上,轉而通過各種金屬離子摻雜[84, 85]、非金屬離子修飾[86]、與石墨烯復合等手段提高析氫性能。制得的催化劑與碳布是一個整體,無需粘合劑,節約成本,因此將廉價、導電性和穩定性優異的碳布用于析氫反應,可望實現未來大規模的氫能源開發。
孫旭平課題組在利用碳布為基底的析氫電極材料方面做了大量的研究工作[87-89]。Tian等[27]在碳布(CC)上生長了Co(OH)F納米線陣列,再低溫磷化得到自支撐多孔CoP納米線陣列(CoP/CC)。這種三維CoP/CC結構規整,可直接用作電化學析氫陰極,在0~14 的pH值范圍內顯示出優異的催化活性,與目前性能最優的Pt/C活性差異較小,經過8000 s持續電解,依然保持很好的穩定性。Ren等[90]通過兩步法在碳布基上得到FeMoS4納米桿狀陣列(FeMoS4NRA/CC),在中性條件下展示了優異的電催化活性,電流密度為10 mA/cm2的過電位為204 mV,Tafel斜率為128 mV/dec。Xing等[91]在碳布上水熱生長WO3,在氨氣氛圍下氮化得到氮化鎢(WN/CC)前驅體,最后再電沉積鎳在前驅體上,得到鎳修飾(Ni-WN/CC)的自支撐電極。在堿性條件下,電流密度為10 mA/cm2時的過電位為74 mV,Tafel斜率為71 mV/dec,良好的析氫性能主要歸功于金屬鎳和氮化鎢的協同效應,同樣的金屬離子(Pt)摻雜協同增強電催化性能也被Xing等[92]在最近的研究中報道過。
非金屬離子修飾對材料的析氫性能也有很大的提高。謝毅院士課題組的Chen等[93]報道了N修飾CoS2納米線陣列析氫的工作,采用簡單的固相法原位生長Co(OH)2/CC前驅體,以硫脲作為氮源和硫源,在氬氣保護下得到碳布基支撐的N原子修飾CoS2多孔納米線陣列(N-CoS2NW/CC),析氫機理如圖3a中的示意圖所示。作者對比了沒有N修飾的CoS2納米線陣列(CoS2NW/CC)的析氫活性,在酸性條件下CoS2NW/CC和N-CoS2NW/CC,在電流密度達到50 mA/cm2時的過電位分別為152 mV和245 mV(圖3b),Tafel斜率分別為58 mV/dec和90 mV/dec,非金屬N修飾顯著增加了材料的催化性能,可能的原因是N與C之間電負性差改變了電極表面的多孔形貌結構,導致更多活性位點的暴露,在提高導電率和加速電子轉移的同時,優化氫吸附吉布斯自由能,降低過電勢。Zhang等[94]將NiMo合金薄膜脈沖電沉積到商業碳布上,得到前驅體(NiMo/CC),然后在氮氣氛圍下采用射頻放電等離子處理前驅體,得到黑色多孔N修飾(NiMoN/CC)薄膜。作者對比了其余兩種材料(MoON/CC和Ni3N/CC)的析氫活性,發現在堿性條件下,NiMoN/CC的起始電位(50 mV)明顯小于后兩者的95 mV和161 mV。NiMoN/CC在電流密度為10 mA/cm2的過電位為109 mV,也明顯小于后兩者的146 mV和208 mV,在掃描速率為1 mV/s的條件下三者的Tafel斜率分別為:95 mV/dec,101 mV/dec,123 mV/dec,由此可以看出雙金屬氮化物之間的協同效應促進了材料性能的提高。

圖3 N-CoS2 NW/CC析氫機理示意圖(a),析氫極化曲線(b)[93]Fig.3 Schematic mechanism of hydrogen evolution of N-CoS2 NW/CC (a), HER polarization curves (b) [93]
將碳布與石墨烯結合或對其進行預處理也是研究熱點之一。Li等[95]利用微波等離子體增強化學氣相沉積法,在碳布上垂直生長石墨烯納米片(VAGNs),通過在VAGNs上電沉積FeOOH,低溫磷化得到FeP納米纖維陣列(FeP NRs/VAGNs/CC))。在酸性條件下,該材料具有優異的電化學析氫性能,初始過電位為19 mV,電流密度為10 mA/cm2的過電位僅為53 mV,Tafel斜率42 mV/dec,且展現了長時間的析氫穩定性,垂直排列的石墨烯不僅為FeP的生長提供了三維結構,增加了其表面活性位點,同時提高了電子轉移速率,優化了析氫性能。Lai等[96]通過將碳布進行預氧化處理,在其上生長氮、磷、氧摻雜的多孔石墨碳,得到產物ONPPGC/OCC,10 mA/cm2電流密度下的全水解電壓為1.66 V,同時,在中性和酸性條件下也有良好的催化性能。
3.2.2 碳纖維紙襯底
碳纖維紙是由直徑約8 μm的碳纖維和粘合劑制成,因其具有均勻而致密的孔結構、良好的機械強度、優異的電子傳輸能力而被用于電催化研究中。過渡金屬硫族化合物在這方面的研究比較多[97, 98]。崔屹課題組的Kong等[28]制備了碳紙支撐的CoSe2納米粒子(CoSe2/CFP)三維立體電極,并顯示了優秀的析氫活性和穩定性。作者還討論了CoSe2納米粒子在不同電極上的析氫情況,CoSe2/玻碳電極(GCE)析氫起始電位為200 mV,而CoSe2/CFP電極的起始電位僅為100 mV,同時CoSe2/CFP電極Tafel斜率為42.1 mV/dec,100 mA/cm2電流密度下的過電勢僅為180 mV,這一研究為電解水的大規模工業應用提供了理論基礎和實驗依據。王雙印課題組的Ouyang等[99]首次報道了碳纖維紙支撐的磷摻雜CoS2納米片陣列(P-CoS2/CFP)的制備方法。P-CoS2/CFP顯示出優異的析氫反應活性,起始電位約25 mV,明顯低于無摻雜CoS2的析氫起始電位(50 mV),電流密度達到10 mA/cm2時需過電位67 mV,而CoS2則需要97 mV。P-CoS2/CFP的Tafel斜率為50 mV/dec,而CoS2的Tafel斜率為58 mV/dec。相比于其它鈷基催化劑,P-CoS2/CFP仍是優秀的析氫電催化劑之一。
除了上面提到的硫族化合物以外,還有很多生長在碳纖維紙上的復合物催化劑,也表現出了優異的催化性能[100-103]。Zhao等[104]創新性地報道了支撐在碳紙上的垂直MoS2/MoC納米片,以無水鉬酸鈉、硫脲作為鉬源和硫源,水熱制備了碳紙支撐的MoS2/CC前驅體,并在氣氛爐中750 ℃滲碳處理,得到Mo2C修飾的MoS2目標產物(MoS2/Mo2C/CFP),滲碳過程及條件如圖4a。作者討論了不同滲碳時間對催化性能的影響,發現25 min處理的樣品性能最好,在0.5 M H2SO4條件下,達到10 mA/cm2時僅需過電位67 mV(圖4b),Tafel斜率為53 mV/dec(圖4c),法拉第效率為98%。第一性原理計算也表明:多個硫、氮配位的鉬催化活性位點的氫吸附自由能接近零,這是該催化劑具有優異催化性能的重要原因。An等[105]采用水熱和低溫硫化兩步法,成功在碳紙上將CuS沉積在NiCo2O4納米纖維陣列上制得CuS/NiCo2O4/CFP。在酸性條件下,具有超長(50 h)的析氫穩定性,Tafel斜率僅為41 mV/dec,符合Volmer-Heyrovsky析氫反應動力學假設。作者還利用其進行了全水解測試,在18 mA/cm2的電流密度下,全水解電壓僅為1.5 V。

圖4 MoS2/Mo2C/CFP合成過程示意圖(a),極化曲線(b),Tafel斜率圖(c)[104]Fig.4 Schematic of preparation process of MoS2/Mo2C/CFP (a), polarization curves (b), corresponding Tafel slopes (c) [104]
3.2.3 其它碳材料襯底
除了碳布和碳纖維紙外,其它碳材料如碳纖維墊、碳箔/碳膜以及碳復合薄膜等[106-108],均可以直接用作三維工作電極,無需導電基底和粘接劑,具有良好的析氫效果。
以石墨烯為連接物的自支撐膜電極材料,具有良好的柔韌性、導電性,被廣泛應用在電解水析氫反應中。Grigorieva教授在2013年提出范德華異質結構的概念[109],采用外延生長的方法,以石墨烯為連接物將二維材料如二硫化鉬、二硒化鎢、六方氮化硼等堆積成三維電極,這種弱相互作用制備的催化劑,具有獨特的性質。隨后,Qiao團隊[110]利用該理論制備合成了多孔四氮化三碳納米片層@氮摻雜石墨烯自支撐復合材料(PCN@NG),BET測試表明該催化劑有大的比表面積和孔體積(114 m2/g,0.37 cm3/g)。大的比表面積、充分暴露的催化中心、分層多孔的結構和三維石墨烯導電基底的協同作用,使得PCN@NG薄膜催化劑顯示出可與鉑媲美的優異電催化性能。2015年,Qiao課題組[111]又采用Li插層剝離技術,得到單層WS2薄膜,通過和氧化石墨烯超聲混勻后,在真空輔助下肼和氨氣氛圍下還原得到氮、氧摻雜石墨烯/WS2薄膜,最后在三苯基膦(triphenylphosphane,TPP)和氮氣氛圍下900 ℃煅燒1 h,得到P,N,O共摻雜的石墨烯/WS2薄膜電極材料(WS2@P, N, O-石墨烯薄膜),合成過程如圖5所示。酸性條件下,電流密度10 mA/cm2下需要的過電勢為125 mV (圖5b),Tafel斜率為52.7 mV/dec (圖5c),交換電流密度為0.131 mA/cm2,在電解20 h后依然能夠保持穩定。作者通過探討其析氫催化機理發現,三維分級多孔結構、1T相WS2提供的豐富活性位點以及多雜原子摻雜石墨烯薄膜三者之間的協同作用,大大提高了薄膜電極的析氫性能。
Peng等[112]采用水熱法和真空抽濾法,合成了三維柔性的硫化鈷/還原氧化石墨烯-碳納米管(CoS2/RGO-CNT)復合薄膜電極,研究表明該三維層狀催化劑具有大量的孔結構,高的機械強度和優異的柔性,對催化活性和穩定性起到非常重要的作用。
3.3 三維多孔材料催化電極
前面提到三維金屬襯底、碳材料襯底電極材料雖然能增加電極間氣體的排出,但是其作為導電集流體具有催化活性不高、動力學性能較弱、高電流密度下負載物易脫落以及增加電極質量和成本等特點,限制了其大規模應用。無襯底三維自支撐電極材料,比如納米多孔碳、納米多孔合金等[113, 114],也可以直接作為工作電極,其本身即可作為集流體和催化活性物質,具有制備方法簡單、成本低、機械強度高、穩定性好等特點。三維無襯底材料將成為未來的研究熱點,在大規模析氫領域得到廣泛應用。
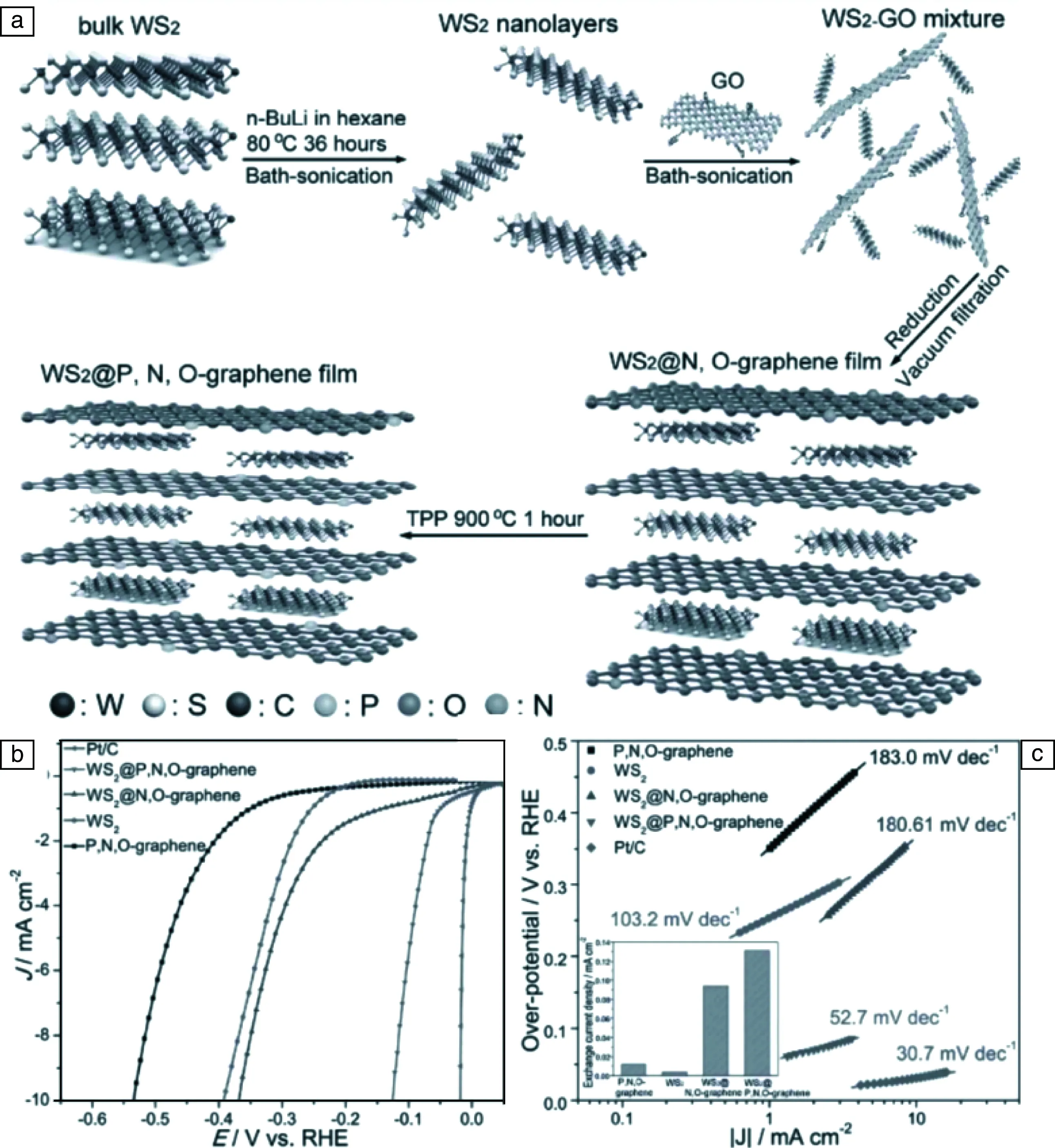
圖5 WS2@P, N, O-石墨烯薄膜合成示意圖(a),極化曲線(b),Tafel斜率圖(插圖為交換電流密度)(c)[111]Fig.5 Schematic of preparation process of WS2@P, N, O-graphene film (a), polarization curves (b), Tafel slopes (inset shows the exchange current density) (c) [111]
模板輔助法是制備三維多孔自支撐催化劑一種常用的手段。2012年,Jaramillo課題組[115]以雙螺旋(double gyroid,DG)多孔硅做模板,在其上電沉積金屬Mo,隨后經過硫化、氫氟酸刻蝕步驟,得到DG構型的高度有序介孔MoS2薄膜催化劑。該DG-MoS2薄膜催化劑,除了擁有大的比表面積以外,還可通過原子尺度調控催化劑表面形貌,使得雙螺旋構型的MoS2活性位點充分暴露,性能測試結果顯示析氫反應中的Tafel斜率為50 mV/dec。在MoS2材料電催化析氫方面也取得了一系列的研究結果,Tan等[116]通過化學去合金法得到納米多孔金(nanoporous gold,NPG),隨后在彎曲的NPG孔洞上氣相沉積MoS2薄膜,合成過程及析氫機理示意圖如圖6a和圖6c所示。作者發現MoS2薄膜層數和彎曲應力對析氫性能的影響很大,研究發現單層MoS2(圖6b)的析氫性能最好,其Tafel斜率為46 mV/dec(圖6d)。該三維納米多孔結構具有大的比表面積,可提供高密度的催化活性位點。更重要的是,多孔金屬的三維雙連續彎曲結構的大曲率可誘導MoS2的晶格發生畸變,從而將單層MoS2由半導體態轉化為金屬態,形成導電的催化位點,最終有效提升MoS2的電催化析氫性能。這一研究不僅解決了制備大面積原子層MoS2薄膜的技術難題,同時也加深了人們對彎曲應力誘導提升催化劑催化性能的認識,為進一步設計和調控催化劑的電催化析氫反應提供了新思路和新途徑。Deng等[117]采用商用泡沫聚氨酯(polyurethane,PU)泡沫作為犧牲模板,制得多孔MoP泡沫,其作為析氫電極具有100%的法拉第效率,交換電流密度為3.052 mA/cm2。這一方法為廉價、規模化制備高效析氫催化劑提供了可行性方法。
Chen課題組長期從事納米多孔金屬、非金屬材料的研究[118-120],Ito等[121]采用化學氣相沉積法與犧牲模板法結合的方法合成硫、氮共摻雜多孔石墨烯電極。他們首先對Ni30Mn70薄片進行脫合金處理,得到納米多孔Ni,隨后采用化學氣相沉積法,在納米多孔Ni表面包覆一層硫、氮共摻雜的石墨烯,再經酸處理除掉金屬鎳骨架,得到硫、氮共摻雜多孔石墨烯電極,具體合成步驟及條件如圖7a所示。作者系統研究了不同氣相沉積溫度下N、S雙原子、N/S單原子及無原子摻雜的多孔石墨烯電極,他們發現沉積溫度對產物的催化活性影響較大,在低溫500 ℃時氮硫共沉積的樣品析氫性能最好,Tafel斜率為81 mV/dec(圖7b)。研究發現低溫更有利于N,S形成晶格缺陷,理論計算發現C-S-N三原子的協同效應能顯著降低氫脫附自由能(圖7c),顯示出與二硫化鉬相近的催化活性。同時,Ito等[122]還對多孔石墨烯中的化學摻雜和拓撲缺陷對析氫催化性能影響的關系進行了深入討論,高度彎曲的石墨烯具有更多的幾何拓撲缺陷,優化了局域電子結構,其化學摻雜水平和活性原子密度得到顯著增強,因此拓撲缺陷和化學摻雜的共同作用促進了多孔碳的析氫性能。Qiu等[123]報道了單原子金屬Ni錨定摻雜多孔石墨烯的研究,相比起其它鎳基或石墨烯基電極,該材料表現出優異的催化活性。在0.5 M硫酸條件下,Tafel斜率為45 mV/dec,過電勢為50 mV。理論和實驗結果表明,Ni-C之間sp-d軌道雜化促進了電荷轉移,空的局域雜化軌道促進了催化并提高了長效析氫穩定性。

圖6 MoS2@NPG合成示意圖(a),單層MoS2@NPG高角環形明場掃描透射電鏡照片(b),單層MoS2@NPG析氫機理示意圖(c), Tafel斜率圖(d)[116]Fig.6 Schematic of preparation process of MoS2@NPG (a), HAABF-STEM image of monolayer MoS2@NPG (b), schematic mechanism of hydrogen evolution of MoS2@NPG HER (c), corresponding Tafel slopes (d) [116]
納米多孔合金材料,因其具有連續納米多孔結構,故在增強材料電子傳輸性、增大比表面積、暴露更多活性位點的同時,可以促進氣體擴散和傳質,提高反應析氫性能。早在2001年,Erlebacher等[124]就試圖通過建立理論模型去揭示脫合金過程中納米多孔金材料形成的內在物理機制,以拓寬其在生物傳感方面的應用。但是,由于多孔金價格昂貴,故在實際使用中受到限制。過渡金屬化合物近年來受到人們廣泛關注,有望成為替代貴金屬的新材料之一。將三維納米多孔金屬材料直接用于催化析氫領域的報道較少,研究者們在這一方面做了大量研究工作。針對傳統去合金法得到的多孔化合物易碎不穩定的特性,Tan等[125]創新性地采用電弧熔融純鈷和磷化鈷得到Co80P20(at%)母合金錠,隨后通過熔融紡絲技術得到柔性光亮的Co2P合金帶,最后利用電化學刻蝕技術,選擇性溶解不穩定的六角密堆積金屬鈷相,得到納米多孔Co2P合金帶,合成路線及相應樣品實物圖如圖8a所示。研究表明,雙連續開放多孔結構的Co2P合金帶(表面多孔形貌如圖8b),當孔徑約為30 nm時,在酸性和堿性條件下均具有優異的析氫催化活性,尤其是在堿性條件下,初始過電勢僅為15 mV,10 mA/cm2下需要的過電勢僅為60 mV,當電流密度超過23 mA/cm2時,析氫過電勢小于Pt/C催化劑(圖8c),np-Co2P電極Tafel斜率(40 mV/dec)也遠遠小于相同條件下的Pt/C電極的Tafel斜率(51 mV/dec),如圖8d所示。采用同樣的合成方法[30],作者又制備了鐵摻雜鈷鐵雙金屬多孔磷化物((Co1-xFex)2P),通過調節Co/Fe原子比和孔徑尺寸,雙金屬多孔磷化物在酸堿條件下均展現了優異的析氫、析氧活性,特別值得注意的是,將其直接作為全解水雙電極,達到10 mA/cm2電流密度時的分解電壓僅為1.53 V。鐵元素的摻雜、鈷鐵之間的協同效應大大降低了CoP的氫吸附自由能,提高了催化活性。上述研究為制備傳統去合金法無法合成的多孔金屬化合物(如:金屬硫化物、金屬氮化物、金屬碳化物、金屬氧化物)和雙金屬化合物提供了借鑒意義,有望取代貴金屬Pt/Ru/Ir等在大規模電解水中得到應用。
圖7 氮、硫共摻雜納米多孔石墨烯合成示意圖(a),不同樣品的Tafel斜率圖(b),氫吸附自由能圖(c)[121]Fig.7 Schematic of preparation process of N and S co-doped nanoporous graphene (a), Tafel slopes of different samples (b), diagram of calculated HER free energy with different samples (c) [121]

圖8 np-Co2P合成示意圖及對應照片(a),掃描電鏡照片(b),堿性條件下的極化曲線(c),Tafel斜率圖(d)[125]Fig.8 Schematic and optical photographs of preparation process of np-Co2P (a), SEM image of np-Co2P (b), polarization curves (c), Tafel slopes in 1.0 M KOH (d) [125]
綜上所述,雖然三維多孔材料在析氫領域的報道和應用相對較少,但因其獨特的物理、化學性質以及顯示出的優異析氫催化性能,必將在未來的大規模能源存儲與轉化方面得到重要應用。
4 結 語
本文綜述了近年來三維自支撐電極材料在電解制氫方面的進展,包括三維金屬材料、三維碳材料以及三維多孔材料等。三維自支撐電極材料本身具有良好的導電性、豐富互通的孔道結構和大的比表面積,有利于電荷轉移和氣體物質傳輸。此外,由于其機械強度較好、無需額外粘接劑,因此具有良好的應用前景。
在當前報道的三維自支撐電極中,很多材料都表現出與鉑十分接近或超過鉑的催化效率,且從實驗和理論角度對催化劑的設計和催化機理認識不斷深入[126],探索新型非貴金屬三維自支撐功能電極在未來的主要挑戰和發展方向主要有以下幾個方面:① 三維材料的結構和形貌對催化性能影響較大,探索合成三維有序陣列或特定表面形貌的催化劑是一個重要研究方向。② 建立大尺度理論模型、借助原位表征等技術手段,理論指導實踐,定性和定量探討三維結構對催化機理的影響,值得深入探索。③ 以高效三維自支撐催化電極為基礎,針對性地優化電解裝置、隔膜和電解液,特別是在堿性和中性電解液條件下,深入挖掘其電解析氫性能,推進功能電極的應用研究。④ 拓展析氫三維自支撐電極的應用領域,研究其在析氧反應和氧還原反應等多功能催化的性能與機理,發展復合多功能催化電極。
參考文獻 References
[1] Gray H B.NatureChemistry[J], 2009, 1 (1): 7.
[2] Kummerer K.AngewandteChemieInternationalEdition[J], 2017, 56: 2-4.
[3] Turner J A.Science[J], 2004, 305 (5686): 972-974.
[4] Upham D C, Agarwal V, Khechfe A,etal.Science[J], 2017, 358: 917-921.
[5] Walter M G , Warren E L, McKone J R ,etal.ChemicalReviews[J], 2010, 110 (11): 6446-6473.
[6] Cook T R , Dogutan D K , Reece S Y,etal.ChemicalReviews[J], 2010, 110 (11): 6474-6502.
[7] Nocera D G.AccountsofChemicalResearch[J], 2012, 25 (5): 767-776.
[8] McKone J R, Lewis N S, Gray H B.ChemistryofMaterials[J], 2013, 26 (1): 407-414.
[9] Subbaraman R, Tripkovic D, Chang K C,etal.NatureMaterials[J], 2012, 11 (6): 550-557.
[10] Lv Z, Mahmood N, Tahir M,etal.Nanoscale[J], 2016, 8 (43): 18250-18269.
[11] Chaudhari N K, Jin H, Kim B,etal.Nanoscale[J], 2017, 9 (34): 12231-12247.
[12] Zhao H, Zhu Y P, Yuan Y Z.EuropeanJournalofInorganicChemistry[J], 2016, 2016 (13-14): 1916-1923.
[13] Tan C L, Cao X, Wu X J,etal.ChemicalReviews[J], 2017, 117 (9): 6225-6331.
[14] Liu K H, Zhong H X, Li S J,etal.ProgressinMaterialsScience[J], 2018, 92: 64-111.
[15] Vikraman D, Akbar K, Hussain S,etal.NanoEnergy[J], 2017, 35: 101-114.
[16] Guo B J, Yu K, Li H L,etal.ACSApplMaterInterfaces[J], 2017, 9 (4): 3653-3660.
[17] Pu Z H, Wang M, Kou Z K,etal.ChemicalCommunications[J], 2016, 52 (86): 12753-12756.
[18] Li F, Li J, Cao Z,etal.JournalofMaterialsChemistryA[J], 2015, 3 (43): 21772-21778.
[19] Yang L J, Yu J Y, Wei Z Q,etal.NanoEnergy[J], 2017, 41: 772-779.
[20] Lin Y, Liu M, Pan Y,etal.JournalofMaterialsScience[J], 2017, 52 (17): 10406-10417.
[21] Liao L, Wang S, Xiao J J,etal.EnergyEnvironmentalScience[J], 2014, 7 (1): 387-392.
[22] Xiao P, Ge X M, Wang H B,etal.AdvancedFunctionalMaterials[J], 2015, 25 (10): 1520-1526.
[23] Kuang P Y, Tong T, Fan K,etal.ACSCatalysis[J], 2017, 7 (9): 6179-6187.
[24] Ma F X, Wu H B, Xia B Y,etal.AngewandteChemie,InternationalEdition[J], 2015, 54 (51): 15395-9.
[25] Yin Y, Han J, Zhang Y,etal.JournaloftheAmericanChemicalSociety[J], 2016, 138 (25): 7965-72.
[26] Zhang J, Wang Q, Wang L,etal.Nanoscale[J], 2015, 7 (23): 10391-7.
[27] Tian J, Liu Q, Asiri A M,etal.JournaloftheAmericanChemicalSociety[J], 2014, 136 (21): 7587-7590.
[28] Kong D S, Wang H T, Lu Z Y,etal.JournaloftheAmericanChemicalSociety[J], 2014, 136 (13): 4897-4900.
[29] Zhang J, Wang T, Liu P,etal.NatureCommunications[J], 2017, 8: 15437.
[30] Tan Y W, Wang H, Liu P,etal.EnergyEnvironmentalScience[J], 2016, 9 (7): 2257-2261.
[31] Morales-Guio C G, Stern L A, Hu X L.ChemicalSocietyReviews[J], 2014, 43 (18): 6555-69.
[32] Wang Y, Kong B, Zhao D Y,etal.Nanotoday[J], 2017, 15: 26-55.
[33] Wang J, Xu F, Jin H Y,etal.AdvancedMaterials[J], 2017, 29 (14): 1605838.
[34] Zou X X, Zhang Y.ChemicalSocietyReviews[J], 2015, 44 (15): 5148-5180.
[35] Anantharaj S, Ede S R, Sakthikumar K,etal.ACSCatalysis[J], 2016, 6 (12): 8069-8097.
[36] Ledendecker M, Calderon S K, Papp C,etal.AngewandteChemieInternationalEdition[J], 2015, 54 (42): 12361-12365.
[37] Yoon T, Kim K S.AdvancedFunctionalMaterials[J], 2016, 26 (41): 7386-7393.
[38] Wang X G, Kolen'ko Y V, Bao X Q,etal.AngewandteChemieInternationalEdition[J], 2015, 54 (28): 8188-8192.
[39] Chang Y H, Lin C T, Chen T Y,etal.AdvancedMaterials[J]. 2013, 25 (5): 756-760.
[40] Zhou H Q, Yu F, Sun J Y,etal.NanoLetters[J], 2016, 16 (12): 7604-7609.
[41] Zhao Z H, Schipper D E, Leitner A P,etal.NanoEnergy[J], 2017, 39: 444-453.
[42] Zhou H Q, Yu F, Sun J Y,etal.JournalofMaterialsChemistryA[J] 2016, 4 (24): 9472-9476.
[43] Zhou H Q, Yu F, Huang Y F,etal.NatureCommunications[J], 2016, 7: 12765.
[44] Zhou H Q, Yu F, Liu Y Y,etal.EnergyEnvironmentalScience[J], 2017, 10 (6): 1487-1492.
[45] Tang T, Jiang W J, Niu S,etal.JournaloftheAmericanChemicalSociety[J], 2017, 139 (24): 8320-8328.
[46] Tang C, Cheng N Y, Pu Z H,etal.AngewandteChemieInternationalEdition[J], 2015, 54 (32): 9483-9487.
[47] Zhang Y, Liu Y W, Ma M,etal.ChemicalCommunications[J], 2017, 53 (80): 11048-11051.
[48] You B, Liu X, Hu G X,etal.JournaloftheAmericanChemicalSociety[J], 2017, 139 (35): 12283-12290.
[49] Geng X R, Wu W, Li N,etal.AdvancedFunctionalMaterials[J], 2014, 24 (39): 6123-6129.
[50] Yan X D, Tian L H, He M,etal.NanoLetters[J], 2015, 15 (9): 6015-6021.
[51] Chen Y Y, Zhang Y, Zhang X,etal.AdvancedMaterials[J], 2017, 29(39): 1703311.
[52] Sivanantham A, Ganesan P, Shanmugam S.AdvancedFunctionalMaterials[J], 2016, 26 (26): 4661-4672.
[53] Xiao C L, Li Y B, Lu X Y,etal.AdvancedFunctionalMaterials[J], 2016, 26 (20): 3515-3523.
[54] You B, Jiang N, Sheng M L,etal.ACSCatalysis[J], 2015, 6 (2): 714-721.
[55] Chen P Z, Zhou T P, Zhang M X,etal.AdvancedMaterials[J], 2017, 29 (30).
[56] Chen G F, Ma T Y, Liu Z Q,etal.AdvancedFunctionalMaterials[J], 2016, 26 (19): 3314-3323.
[57] Feng L L, Yu G T, Wu Y Y,etal.JournaloftheAmericanChemicalSociety[J], 2015, 137 (44): 14023-14026.
[58] Wu Y Y, Li G D, Liu Y P,etal.AdvancedFunctionalMaterials[J], 2016, 26 (27): 4839-4847.
[59] Wu Y Y, Liu Y P, Li G D,etal.NanoEnergy[J], 2017, 35: 161-170.
[60] Liu B, Zhao Y F, Peng H Q,etal.AdvancedMaterials[J], 2017, 29 (19): 1606521.
[61] Tian J Q, Liu Q, Cheng N Y,etal.AngewandteChemieInternationalEdition[J], 2014, 53 (36): 9577-9581.
[62] Yang C, Gao M Y, Zhang Q B,etal.NanoEnergy[J], 2017, 36: 85-94.
[63] Liang Y H, Liu Q, Luo Y L,etal.ElectrochimicaActa[J], 2016, 190: 360-364.
[64] Popczun E J, Read C G, Roske C W,etal.AngewandteChemieInternationalEdition[J], 2014, 53 (21): 5427-5430.
[65] Peng Z, Jia D S, Al-Enizi A M,etal.AdvancedEnergyMaterials[J], 2015, 5 (9): 1402031.
[66] Barman B K, Das D, Nanda K K.JournalofMaterialsChemistryA[J], 2017, 5 (34): 18081-18087.
[67] Balogun M S, Qiu W T, Huang Y C,etal.AdvancedMaterials[J], 2017, 29 (34): 1702095.
[68] Chen Z B, Cummins D, Reinecke B N,etal.NanoLetters[J], 2011, 11 (10): 4168-4175.
[69] Lukowski M A, Daniel A S, English C R,etal.EnergyEnvironmentalScience[J], 2014, 7 (8): 2608-2613.
[70] Faber M S, Dziedzic R, Lukowski M A,etal.JournaloftheAmericanChemicalSociety[J], 2014, 136 (28): 10053-10061.
[71] Yang Y, Fei H , Ruan G D,etal.AdvancedMaterials[J], 2015, 27 (20): 3175-3180.
[72] Liu T T, Liu D N, Qu F L,etal.AdvancedEnergyMaterials[J], 2017, 7 (15): 1700020.
[73] Tang C, Zhang R, Lu W B,etal.AdvancedMaterials[J], 2017, 29 (2): 1602441.
[74] Jiang P, Liu Q, Liang Y H,etal.AngewandteChemieInternationalEdition[J], 2014, 53 (47): 12855-12859.
[75] Pu Z H, Liu Q, Jiang P,etal.ChemistryofMaterials[J], 2014, 26 (15): 4326-4329.
[76] Lu W B, Liu T T, Xie L S,etal.Small[J], 2017, 13 (32): 1700805.
[77] Liu Y P, Li Q J, Si R,etal.AdvancedMaterials[J], 2017, 29 (13): 1606200.
[78] Yu L, Zhou H Q, Sun J Y,etal.EnergyEnvironmentalScience[J], 2017, 10 (8): 1820-1827.
[79] Bu L Z, Guo S J, Zhang X,etal.NatureCommunications[J], 2016, 7: 11850.
[80] Pi M Y, Wu T L, Zhang D K,etal.Nanoscale[J], 2016, 8 (47): 19779-19786.
[81] Chen P G, Xu K, Tao S,etal.AdvancedMaterials[J], 2016, 28 (34): 7527-7532.
[82] Fan M H, Chen H, Wu Y Y,etal.JournalofMaterialsChemistryA[J]. 2015, 3 (31): 16320-16326.
[83] Feng L L, Fan M H, Wu Y Y,etal.JournalofMaterialsChemistryA[J], 2016, 4 (18): 6860-6867.
[84] Miao J W, Xiao F X, Yang H B,etal.ScienceAdvance[J], 2015, 1 (7): e1500259.
[85] Zhang Z, Liu Y D, Ren L,etal.ElectrochimicaActa[J], 2016, 200: 142-151.
[86] Wang X D, Xu Y F, Rao H S,etal.EnergyEnvironmentalScience[J], 2016, 9 (4): 1468-1475.
[87] Jiang P, Liu Q, Sun X P.Nanoscale[J], 2014, 6 (22): 13440-13445.
[88] Liu Q, Shi J L, Hu J M,etal.ACSAppliedMaterialsInterfaces[J], 2015, 7 (7): 3877-3881.
[89] Liu Q, Xie L S, Qu F L,etal.InorganicChemistryFrontiers[J], 2017, 4 (7): 1120-1124.
[90] Ren X, Wang W Y, Ge R X,etal.ChemicalCommunications[J], 2017, 53 (64): 9000-9003.
[91] Xing Z C, Wang D W, Li Q,etal.ElectrochimicaActa[J], 2016, 210: 729-733.
[92] Xing Z C, Han C, Wang D W,etal.ACSCatalysis[J], 2017, 7 (10): 7131-7135.
[93] Chen P Z, Zhou T P, Chen M L,etal.ACSCatalysis[J], 2017, 7(11): 7405-7411.
[94] Zhang Y Q, Ouyang B, Xu J,etal.AdvancedEnergyMaterials[J], 2016, 6 (11): 1600221.
[95] Li D Q, Liao Q Y, Ren B W,etal.JournalofMaterialsChemistryA[J], 2017, 5 (22): 11301-11308.
[96] Lai J P, Li S P, Wu F X,etal.EnergyEnvironmentalScience[J], 2016, 9 (4): 1210-1214.
[97] Zhuo J Q, Cabán-Acevedo M, Liang H F,etal.ACSCatalysis[J], 2015, 5 (11): 6355-6361.
[98] Caban-Acevedo M, Stone M L, Schmidt J R,etal.NatureMaterials[J], 2015, 14 (12): 1245-1251.
[99] Ouyang C B, Wang X, Wang S Y.ChemicalCommunications[J], 2015, 51 (75): 14160-14163.
[100]Wang H T, Kong D S, Johanes P,etal.NanoLetters[J], 2013, 13 (7): 3426-3433.
[101]Zhang L, Wang T, Sun L,etal.JournalofMaterialsChemistryA[J], 2017, 5 (37): 19752-19759.
[102]Ekspong J, Sharifi T, Shchukarev A,etal.AdvancedFunctionalMaterials[J], 2016, 26 (37): 6766-6776.
[103]Zhong X B, Wang J, Zhong H X,etal.ACSNano[J], 2016, 10 (2): 2342-2348.
[104]Zhao Z H, Qin F, Kasiraju S,etal.ACSCatalysis[J], 2017, 7 (10): 7312-7318.
[105]An L, Huang L, Zhou P P,etal.AdvancedFunctionalMaterials[J], 2015, 25 (43): 6814-6822.
[106]Duan J J, Chen S, Jaroniec M,etal.ACSCatalysis[J], 2015, 5 (9): 5207-5234.
[107]Hou Y, Lohe M R, Zhang J,etal.EnergyEnvironmentalScience[J], 2016, 9 (2): 478-483.
[108]Wang X Y, Gan X, Hu T,etal.AdvancedMaterials[J], 2017, 29 (4): 1603617.
[109]Geim A K, Grigorieva I V.Nature[J], 2013, 499 (7459): 419-425.
[110]Duan J J, Chen S, Jaroniec M,etal.ACSNano[J], 2015, 9 (1): 931-940.
[111]Duan J J, Chen S, Chambers B A,etal.AdvancedMaterials[J], 2015, 27 (28): 4234-4241.
[112]Peng S J, Li L L, Han X P,etal.AngewandteChemieInternationalEdition[J], 2014, 53 (46): 1-7.
[113]Chowdhury S, Balasubramanian R.ProgressinMaterialsScience[J], 2017, 90: 224-275.
[114]Lu Q, Hutchings G S, Yu W T,etal.NatureCommunications[J], 2015, 6: 6567.
[115]Kibsgaard J, Chen Z B, Reinecke B N,etal.NatureMaterials[J], 2012, 11 (11): 963-970.
[116]Tan Y W, Liu P, Chen L Y,etal.AdvancedMaterials[J], 2014, 26 (47): 8023-8028.
[117]Deng C, Ding F, Li X Y,etal.JournalofMaterialsChemistryA[J], 2016, 4 (1): 59-66.
[118]Ito Y, Tanabe Y, Qiu H J,etal.AngewandteChemieInternationalEdition[J], 2014, 53 (19): 4922-4926.
[119]Fujita T, Guan P F, McKenna K,etal.NatureMaterials[J], 2012, 11 (9): 775-780.
[120]Ge X B, Chen L Y, Zhang L,etal.AdvancedMaterials[J], 2014, 26 (19): 3100-3104.
[121]Ito Y, Cong W T, Fujita T,etal.AngewandteChemieInternationalEdition[J], 2015, 54 (7): 2131-2136.
[122]Ito Y, Shen Y H, Hojo D,etal.AdvancedMaterials[J], 2016, 28 (48): 10644-10651.
[123]Qiu H J, Ito Y, Cong W T,etal.AngewandteChemieInternationalEdition[J], 2015, 54 (47): 14031-14035.
[124]Erlebacher J, Aziz M J, Karma A,etal.Nature[J], 2001, 410 (6827): 450-453.
[125]Tan Y W, Wang H, Liu P,etal.AdvancedMaterials[J], 2016, 28 (15): 2951-2955.
[126]Seh Z W, Kibsgaard J, Dickens C F,etal.Science[J], 2017, 355 (6321): eaad4998.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
