不飽和聚碳硅烷的研究進展
2018-05-07 07:01:54孫維朋顧竹靈高占臣呂素芳蔣劍雄張飛豹
中國材料進展 2018年3期
孫維朋,顧竹靈,高占臣,呂素芳,蔣劍雄,張飛豹
(杭州師范大學 有機硅化學及材料技術教育部重點實驗室,浙江 杭州 311121)
1 前 言
聚碳硅烷是一類有機硅高分子化合物,其主鏈一般由硅和碳原子交替組成,硅和碳原子上連接有氫或有機基團,分子鏈為線形或支化結構[1]。自Yajima等[2]證明了聚碳硅烷高溫裂解可以制備碳化硅以來,聚碳硅烷的合成和應用逐漸成為研究和應用的熱點。20世紀80年代,國防科技大學提出常壓熱裂解合成聚碳硅烷的新方法[3],聚碳硅烷作為SiC陶瓷基復合材料的前驅體備受關注。此外,聚碳硅烷在生活上的應用十分廣泛,包括污水處理[4]、耐磨涂料[5]、防氧化[6]、抗老化材料[7]、過濾煙爐[8]、無涂料防污漁網[9]等多個領域。不飽和聚碳硅烷是聚碳硅烷的一個重要組成部分[1],當在聚合物主鏈中引入共軛的C=C雙鍵時,高能階Si的σ-成鍵軌道與共軛系統的π*反鍵軌道能夠發生σ-π相互作用[10],尤其當硅硅鏈越長時,在聚合物的主鏈上σ-π共軛現象就越明顯,這種共軛現象拓寬了聚碳硅烷的研究范圍,不僅僅可用于陶瓷前驅體、耐熱材料等,還可以用于光伏材料、電致發光材料、導電材料等,是一類潛力巨大的功能高分子材料。以前對聚碳硅烷的研究綜述主要聚焦于聚碳硅烷的合成方法及其作為SiC陶瓷材料的前驅體的研究。纖維增強SiC陶瓷基復合材料在航空航天、空間技術、能源技術、化工、交通工業等領域具有廣闊的應用前景[11],本文總結了近年來不飽和聚碳硅烷的合成方法,并對其在太陽能電池[12]、發光材料[13]、金屬離子識別[14]等方面的應用進行了綜述。
2 不飽和聚碳硅烷的合成方法
不飽和聚碳硅烷由于結構的特殊性,主鏈上含有共軛基團,合成反應往往需要由特定的單體聚合而成。現有文獻報道的合成方法主要有:硅氫加成法[15]、Heck反應法[16, 17]、Wittig反應法[10]等,其中硅氫加成法是目前合成聚碳硅烷最常用的方法。
2.1 硅氫加成聚合法
2.1.1 硅氫加成反應機理簡介
硅氫加成反應是制備Si-C鍵最重要的反應之一,對許多含有如脂基、腈基、胺基、酰胺、硝基、醚等基團的化合物都能進行反應,并且反應條件溫和。比較常用的催化劑[18]主要有鉑[19]、銠[20]、釕[21]、鎳[22]、鈀[23]等的配合物催化劑,其具有高活性和高選擇性的特點。
最早被廣泛認可的硅氫加成反應機理由Chalk和Harrod提出[24]。這種機理最初源于對氯鉑酸作為Pt催化劑的研究,其為其他過渡金屬絡合物催化劑的作用提供了定性合理的概括,炔烴發生硅氫加成反應機理如圖1所示。
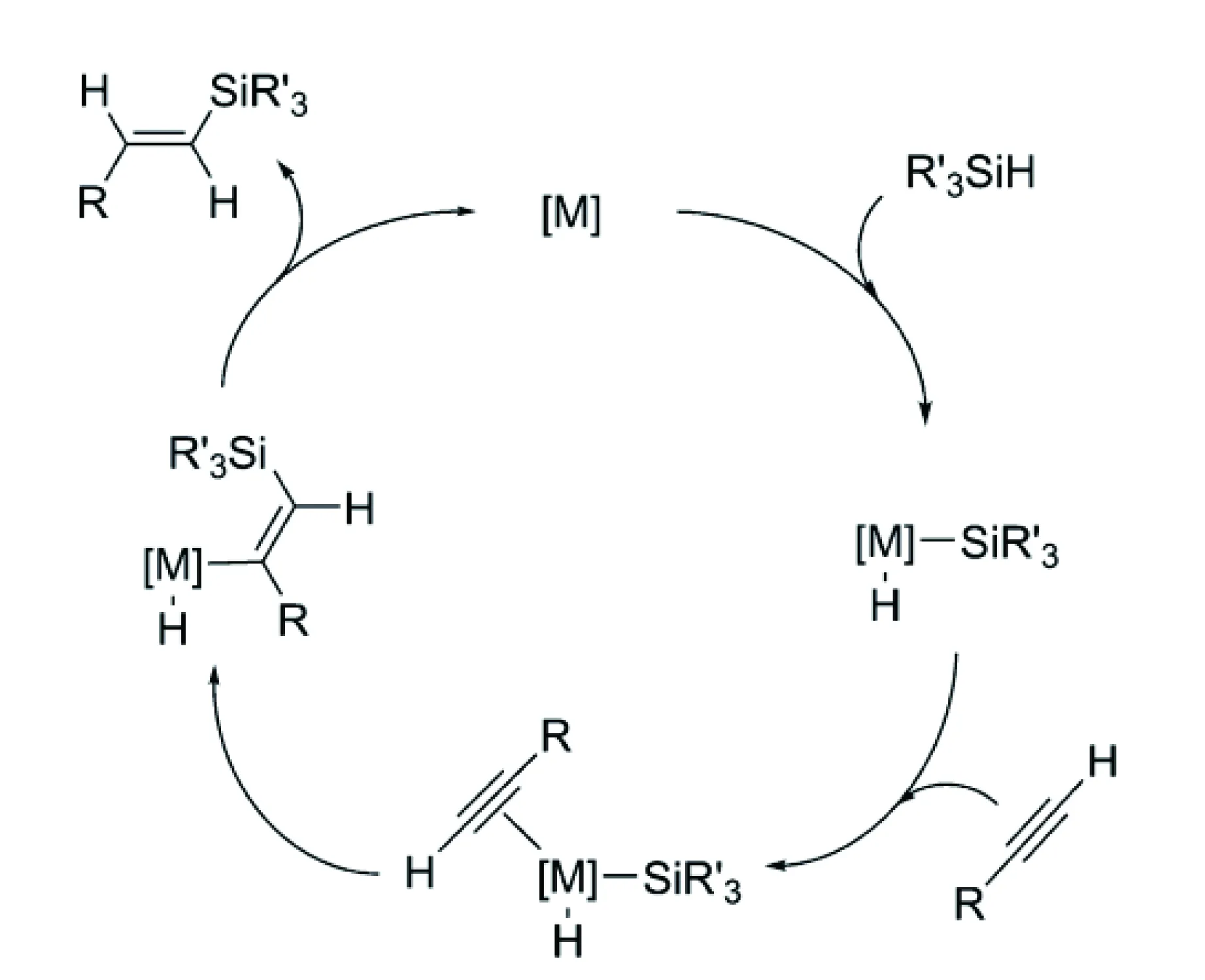
圖1 Chalk和 Harrod所提出的硅氫加成的反應機理[24]Fig.1 The mechanism of hydrosilylation proposed by Chalk and Harrod[24]
隨后,Ojima等[25]提出了一種新的機理(如圖2所示),在遷移插入步驟中,甲硅烷基首先發生金屬化之后,再進行E/Z異構化。由于甲硅烷基和取代的金屬原子之間的立體排斥,(Z)-甲硅烷基亞乙烯基中間體容易異構化為熱力學有利的(E)-甲硅烷基亞乙烯絡合物。
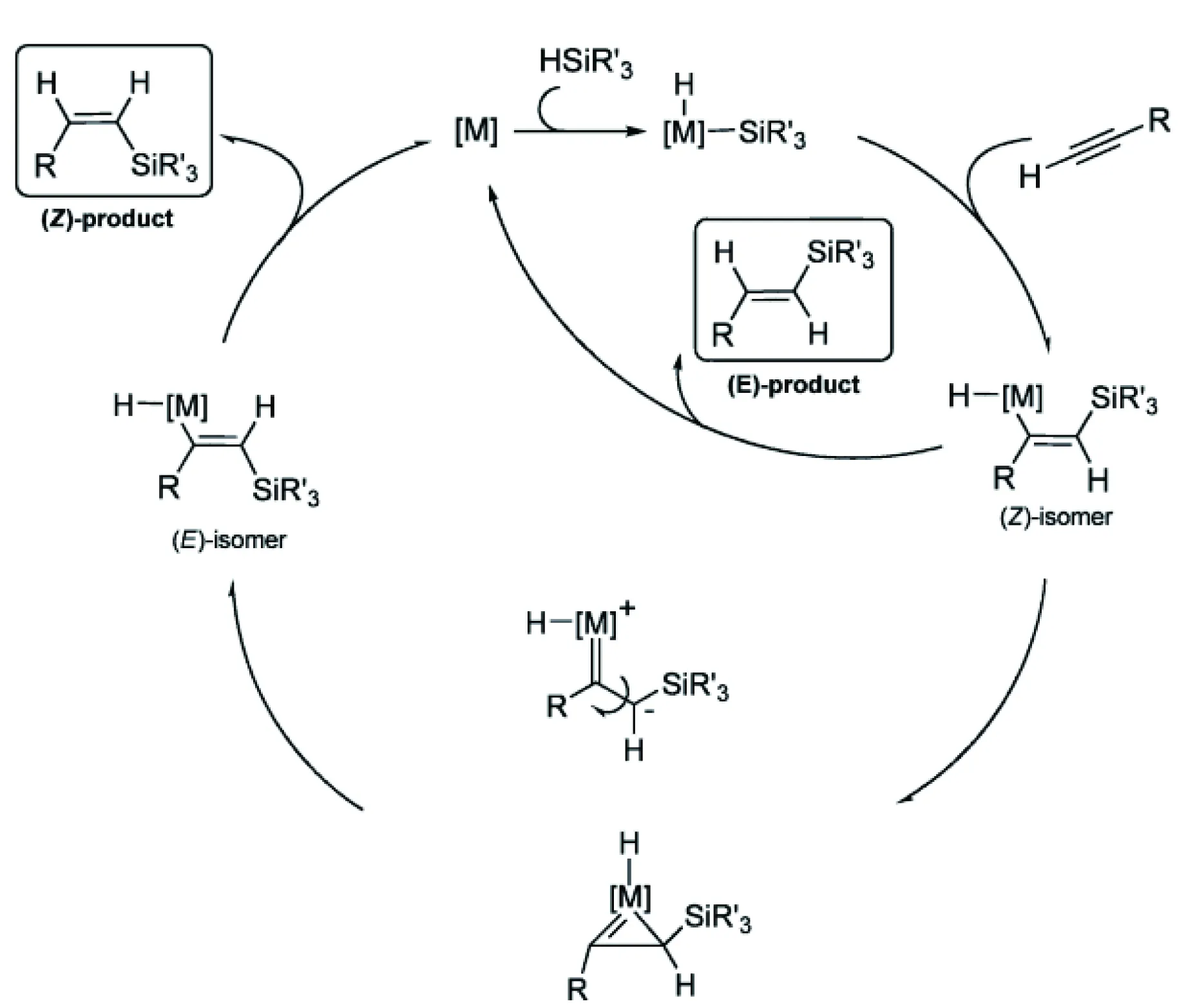
圖2 選擇性炔烴硅氫加成的Crabtree-Ojima機理[25]Fig.2 The Crabtree-Ojima mechanism for hydrosilylation [25]
2.1.2 硅氫加成制備不飽和聚碳硅烷
對炔烴的硅氫加成一般有兩種方式:第一類是同時含有硅氫和炔烴的化合物的自聚反應;第二類是二氫硅烷與二炔烴的共聚反應。
2.1.2.1 同時含有硅氫和炔烴的化合物的自聚反應
Barton等[26]用CPA ( Chloroplatanic acid )催化聚合,得到了均聚的聚合物1,反應式如圖3,這類反應單體和反應的條件比較簡單,得到的聚合物結構也相對規整。
Xiao等[27]報道了類似的硅氫加成反應,得到了超支化聚碳硅烷,顯然硅氫加成反應不僅能用來合成線性的聚合物,還可以合成具有復雜結構的樹狀聚合物。Kim[28]合成了相應化合物的鄰、間、對的構型,并用這些化合物進行自聚反應,發現間位和對位的單體自聚后能產生線性的聚合物,但鄰位的單體不能產生線性的聚合物。另外,單體反應溫度不同,得到的聚合物結構也有差別,比如THF加熱回流下得到的主鏈是由硅亞甲基和碳原子組成的,而常溫條件下得到的是偶聯產物。
在Neckers等[29]的文章中報道了用圖4所示的化合物進行自聚得到聚合物2和3,在這兩個化合物中硅亞甲基和乙炔基是相鄰的,中間不像其它的化合物那樣有芳環分隔開。這樣的化合物硅氫加成自聚得到的產物主鏈是硅亞甲基和乙烯基交替組成的聚合物,相應的芳環成為了以硅碳為主鏈的聚合物上的取代基。這個反應的反應條件與其他反應的條件不同在于使用了輻射波長為350 nm的光反應器(Rayonet reactor)來進行反應。
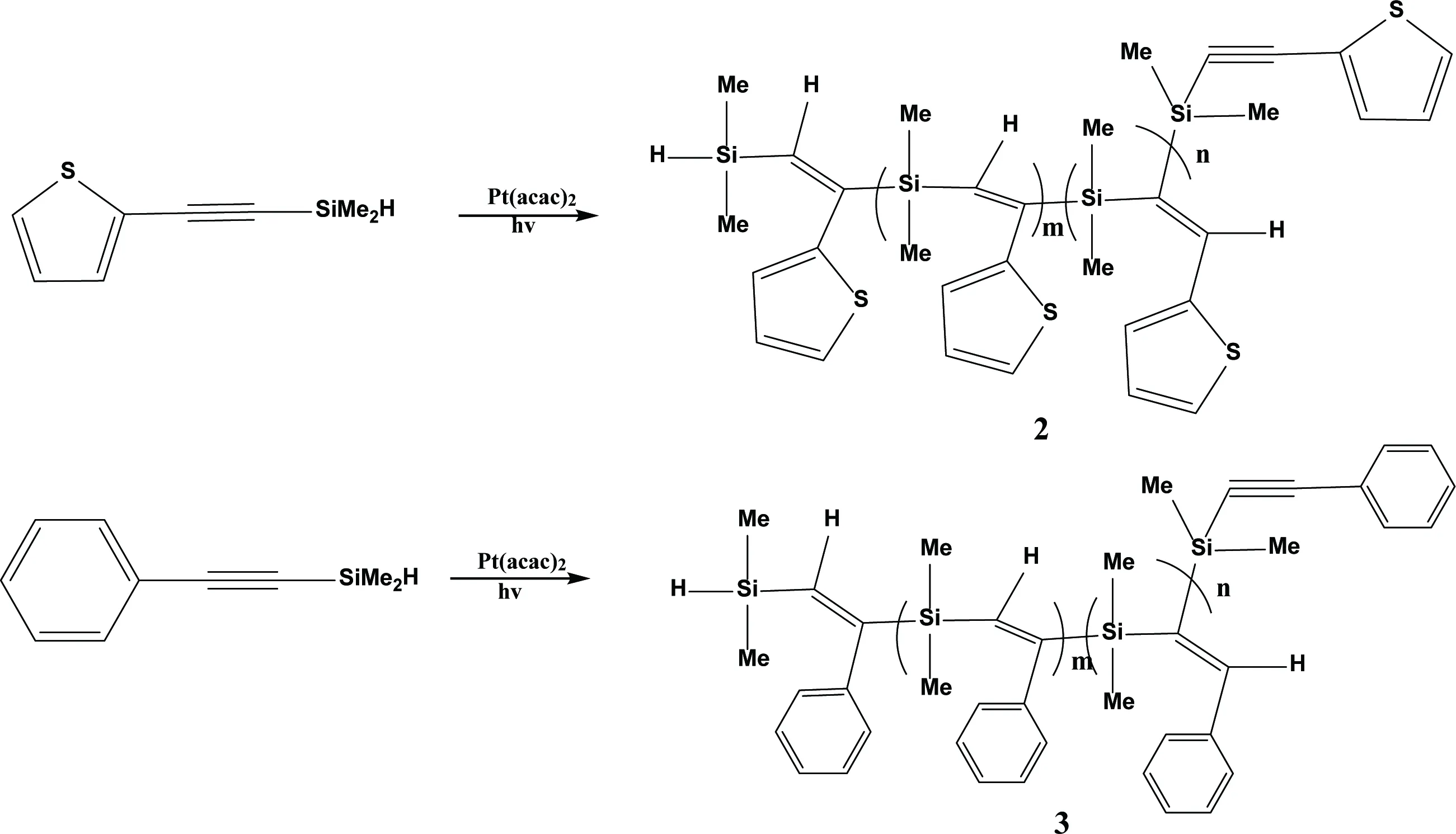
圖4 合成聚碳硅烷[29]Fig.4 Synthesis of polycarbosilane [29]
2.1.2.2 二氫硅烷與二炔烴的共聚反應
Kim等[30]報道了二氫硅烷與二炔烴化合物的硅氫加成反應,這類反應相對比較簡單,反應所需的硅烷可以買到,聚合物的結構通常由硅烷上的取代基結構決定。當硅烷上的取代基為苯基時,主要以β-加成產物為主。Yamashita等[31]報道Pd-PCy3體系可以有效地催化二氫硅烷與對位或間位的二乙炔基苯的硅氫化反應,反應速度較快,熱重分析表明這些化合物具有較好的熱穩定性。2012年,Yamashita等[32]報道了具有亞甲硅烷基-亞乙烯基-亞苯基-亞乙烯基為骨架和三苯胺或含咔唑單元為側鏈的聚碳硅烷可以通過Pd-PCy3催化來制備,并且對其光學和熱力學進行分析,5%的質量損失區間在382 ℃~444 ℃之間,表明此聚合物具有相當高的熱穩定性,可以作為新型的光電材料。
1998年,Mori等[33]報道了RhCl(PPh3)3/NaI催化端炔烴硅氫化反應合成了含硅雜原子取代基的E-和Z-烯基硅烷。在0 ℃下反應2 h生成的是E-式產物,在55 ℃下反應生成的是Z-式產物(如圖5)。
隨后,Mori等[34]在2000年報道了將上述催化劑用來催化雙炔烴化合物和雙硅氫化合物合成高分子聚合物,可以在控制反應條件下得到E-式或Z-式聚合物,并且對合成的過程進行了說明,如果硅氫化合物和炔烴同時加入,控溫60 ℃會得到E-式產物;如果先加入溶劑,催化劑和硅氫化合物在0 ℃下攪拌反應1 h,再加入雙炔烴化合物,反應2 h后升至室溫,并在室溫下繼續反應,最后會得到Z-式的產物。2004年,Mori等[35]發現使用[RhI(cod)]2催化劑時,改變加料順序,也可以控制反應立體構型,得到E-式或Z-式產物。
Kwak等[36]在控制硅氫反應產物構型方面也做了很多工作。2004年報道了用RhI(PPh3)3[37]催化雙炔烴化合物和雙硅氫化合物的反應(如圖6),相應的反應如果在鄰二氯苯溶劑中進行,150~180 ℃下會得到cis-式的產物,而在甲苯溶劑中,0 ℃下,得到的聚合物為trans-式的產物,并且發現在紫外光的照射下產物的構型會由cis-構型轉變成trans-的構型。
Chen等[38]也報道了類似的反應,所使用的硅氫化合物與上文不同,硅亞甲基和芳環之間一般由乙烯基相連。但其單體合成相對比較困難[39],先要合成硫縮醛,再將硫碳鍵斷開形成碳碳雙鍵,最后還原得到目標產物。在此基礎上,他們組合成了一系列的硅乙烯基間隔的給受體交錯的聚合物[40]。

圖5 RhCl(PPh3)3/NaI催化的端炔烴硅氫加成反應[33]Fig.5 Hydrosilylation of terminal alkynes catalyzed by RhCl(PPh3)3/NaI[33]

圖6 在RhI(PPh3)3催化劑下合成cis-和trans-聚合物[37]Fig.6 Synthesis of cis-and trans- polymers by RhI(PPh3)3 [37]
Perry等[41]報道了1,3-雙炔烴化合物與雙硅氫化合物進行反應,得到了cis,cis-2,3-雙取代的1,3-丁二烯的聚合物(如圖7)。
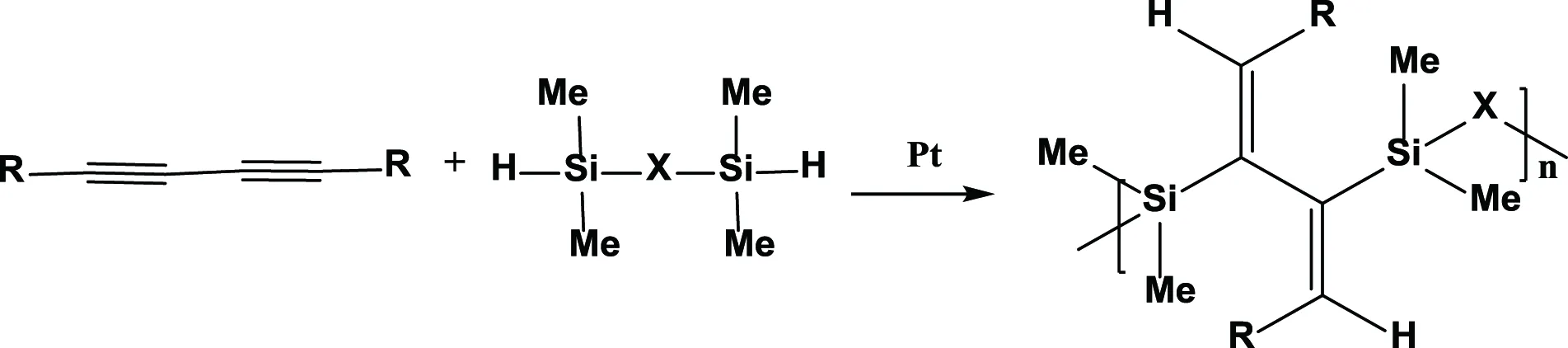
圖7 二炔化合物的硅氫加成反應[41]Fig.7 Hydrosilylation of diynes [41]
Neckers等[29]利用Pt(acac)2光催化劑催化過量的4-二甲基硅基苯乙炔與1,4-對二乙炔基苯(10 ∶1)進行硅氫化反應,生成了具有良好光物理性質的聚合物。
2010年,Sasano等[42]利用1,4-二(3-乙炔基-9-咔唑基)苯與二氫硅烷在含有銠催化劑的甲苯溶液中反應,得到含咔唑的不飽和聚碳硅烷及其各種衍生物。2012年,Zhao等[43]在RhCl(PPh3)3催化劑作用下,將含炔烴的噻咯與含苯、聯苯等芳香官能團的硅氫化合物反應,得到具有很好熱穩定性的聚合物。該聚合物在溶液中顯示弱熒光,但在聚合體和薄膜狀態下熒光較強,顯示出一定的聚集誘導效應。在溶液和聚集態下,檢測了該聚合物對爆炸物的傳感性質,表明該聚合物具有很強的靈敏度,可用于爆炸物檢測。同年,Zhao等[44]在Karstedt’s催化劑作用下,通過TDMSHS與二乙炔基噻咯反應制備了超支化聚合物(hb-SPSV) (如圖8所示),該聚合物具有良好的熒光特性和熱穩定性。其中TDMSHS是首次合成,并且通過X-ray及光譜方法表征了其結構,該超支化聚合物可用于爆炸物檢測。

圖8 通過鉑催化的TDMSHS與5的硅氫加成合成含噻咯的超支化聚(甲硅基亞乙烯基)(hb-SPSV)[44]Fig.8 Synthesis of hyperbranched poly(silylenevinylene)(hb-SPSV) with TDMSHS and diethynylsilole by Karstedt's catalyst [44]
2017年,Bernard等[45]在烯丙基氫化聚碳硅烷(AHPCS)中引入硼元素,發現硼含量高的聚合物熱解后更適合制備致密性好的陶瓷。同年,Ji等[46]制備了含/不含烯丙基的液態超支化聚合碳硅烷(LHBPCS),并在室溫下通過紫外線輻射進行交聯。-Si-H、-Si-H2和烯丙基在紫外線照射過程中,相同紫外線輻射時間后,含有烯丙基的反應物,反應程度較高。可能發生的交聯反應包括硅氫化反應、烯丙基聚合和脫氫偶聯反應等。與傳統的LHBPCS交聯熱處理相比,紫外線輻射具有溫度低、節約時間、提高不飽和基團的利用效率等優點。通過調節烯丙基含量,可以抑制LHBPCS中孔隙的形成并且熱解后可以轉化成無孔SiC陶瓷。另外,紫外線輻射交聯不需要加入引發劑、反應性稀釋劑和其他材料,是合成LHBPCS的良好交聯方法。Zhong等[47]等也通過紫外線輻射法合成出LHBPCS,在1000 ℃熱解得到化學計量比的SiC陶瓷,該SiC陶瓷在空氣中、1400 ℃下依然具有良好的熱穩定性。
2.2 Heck反應制備不飽和聚碳硅烷
Heck反應[16, 17]主要是硅烯烴與鹵代芳烴在Pd催化下反應,偶聯得到聚碳硅烷。圖9所示的反應是利用Heck反應得到在主鏈中含有電子傳輸惡二唑和空穴傳輸咔唑部分的硅基交替不飽和聚碳硅烷,測試了該聚合物的電致發光性質,并利用該聚合物制備了LED器件。
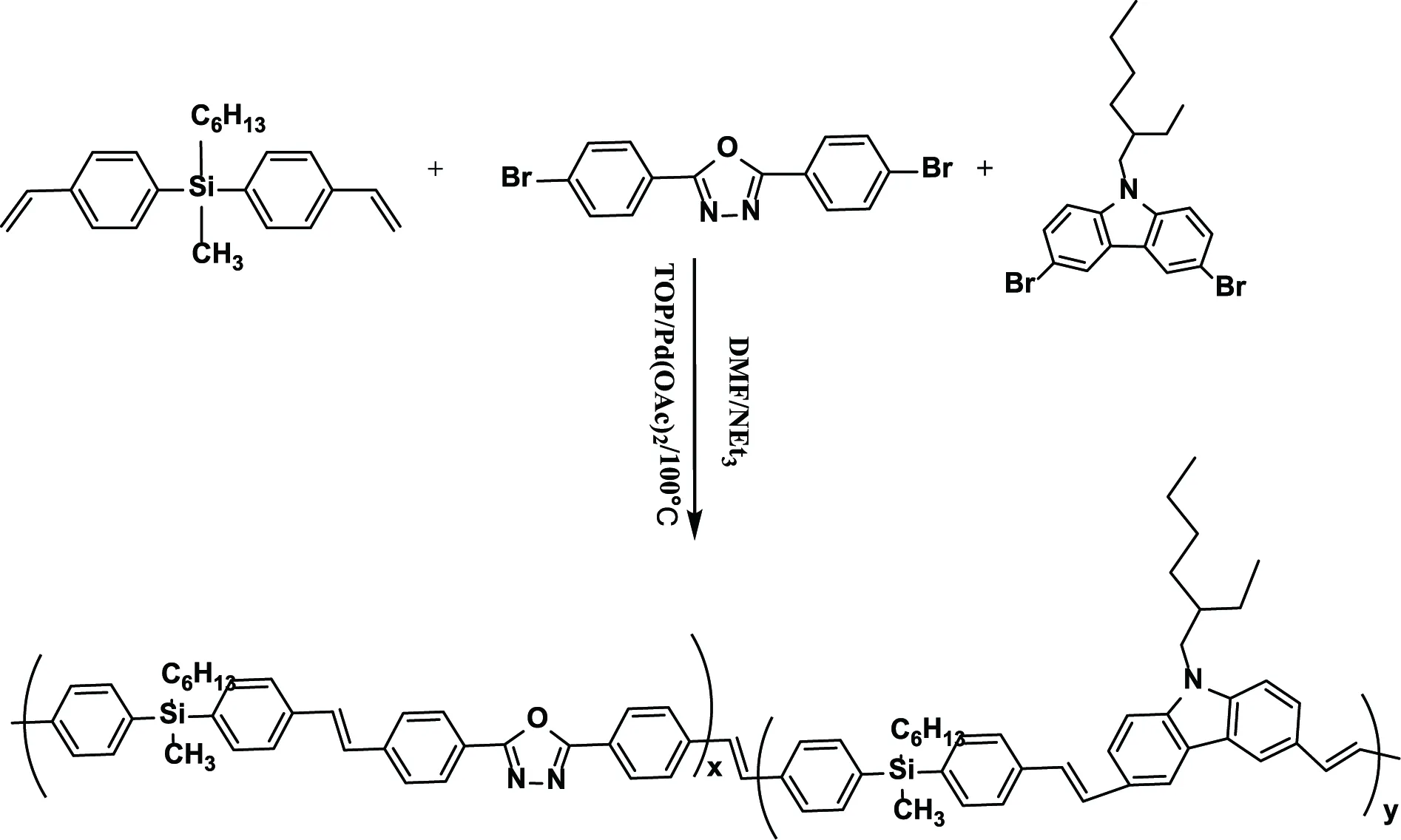
圖9 利用Heck反應合成不同組成的硅基共聚物[16, 17]Fig.9 Synthesis of silicon-containing polymers by Heck reaction [16, 17]
Ma等[48]報道了氯代苝酰亞胺與硅氫化合物,在醋酸鈀和磷配體存在的情況下反應成環,其反應式如圖10所示,該產物相對于原料苝酰亞胺具有明顯的紅移,單晶測量結果表明該產物基本為平面結構,制備成OFET器件的遷移率約0.3 cm2·V-1·s-1。

圖10 一鍋法合成硅雜環戊二烯二酰亞胺[48]Fig.10 Synthesis of sila-annulated perylene diimides by one-pot reaction [48]
2.3 Wittig反應制備不飽和聚碳硅烷
利用Wittig反應[10]制備不飽和聚碳硅烷,相關文獻報道較少,該反應主要是通過磷葉立德與醛反應聚合后得到不飽和聚碳硅烷。Kim等[49]利用含硅的磷鹽與對苯二醛反應,得到硅橋聯的聚對苯乙烯類聚合物,該聚合物可溶于常規的有機溶劑,反應式如圖11所示。
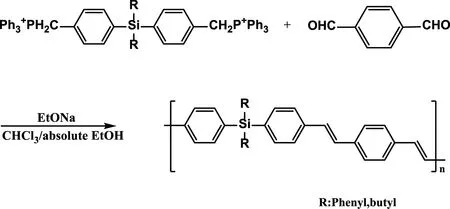
圖11 利用Wittig反應合成不飽和聚碳硅烷[49]Fig.11 Synthesis of unsaturated polycarbosilanes by Wittig reaction [49]
3 不飽和聚碳硅烷的應用
3.1 電化學中的應用
隨著對聚碳硅烷研究的深入,最近發現聚碳硅烷在電化學方面也有良好的性能。這方面的報道比較晚,卻體現了聚碳硅烷有著廣泛應用價值。
近年來,具有碳酸酯結構的聚合物作為固體聚合電解質(SPE)的基體備受關注。Tominaga及其同事報道聚合物骨架中含有碳酸酯基團的聚(碳酸亞乙酯)[50]及其衍生物[51-53]在雙三氟甲烷磺酰亞胺鋰(LiTFSI)的存在下表現出相對高的離子導電率。Mindemark等[54, 55]將柔性的聚碳酸酯作為SPE進行合成和檢測。Grinstaff和同事還合成了聚(乙醚-1,2-甘油碳酸酯)。這些結果表明,具有碳酸酯結構的柔性聚合物是SPE的良好基質。
2012年Matsumoto等[56]制備了具有五元環狀碳酸酯結構(SBMC)的硅雜環丁烷。通過聚合得到聚SBMC,通過線性掃描伏安法檢測聚SBMC的電化學穩定性,發現該聚合物可以用于普通的鋰離子電池。2016年,該課題組[57]又合成了每個重復單元具有一個或兩個五元環狀碳酸酯基團的聚碳硅烷,并通過添加鋰鹽來檢測其作為SPE的特性。發現這些聚合物的離子電導率隨著LiTFSI添加量的增加而增加。這些聚合物/鹽體系可以形成一種“鹽電解質中的聚合物”,表現出相當高的離子電導率。
3.2 太陽能電池給體材料
2014年,Li等[58]通過Suzuki偶聯反應合成以硅芴為電子給體單元、5-烷氧基-6-氟苯并噻二唑為電子受體單元、噻吩為間隔基的(D-A)型交替的共軛聚合物(如圖12)。將其用于聚合物太陽能電池,氟原子的引入可降低聚合物HOMO和LUMO的能級進而提高開路電壓。而在苯并噻二唑單元上引入柔性烷氧基鏈可以增加所得聚合物的溶解性而不干擾聚合物鏈在固態下的緊密堆積。基于PC71BM的聚合物太陽能電池(PSC)轉化效率高達6.41%。
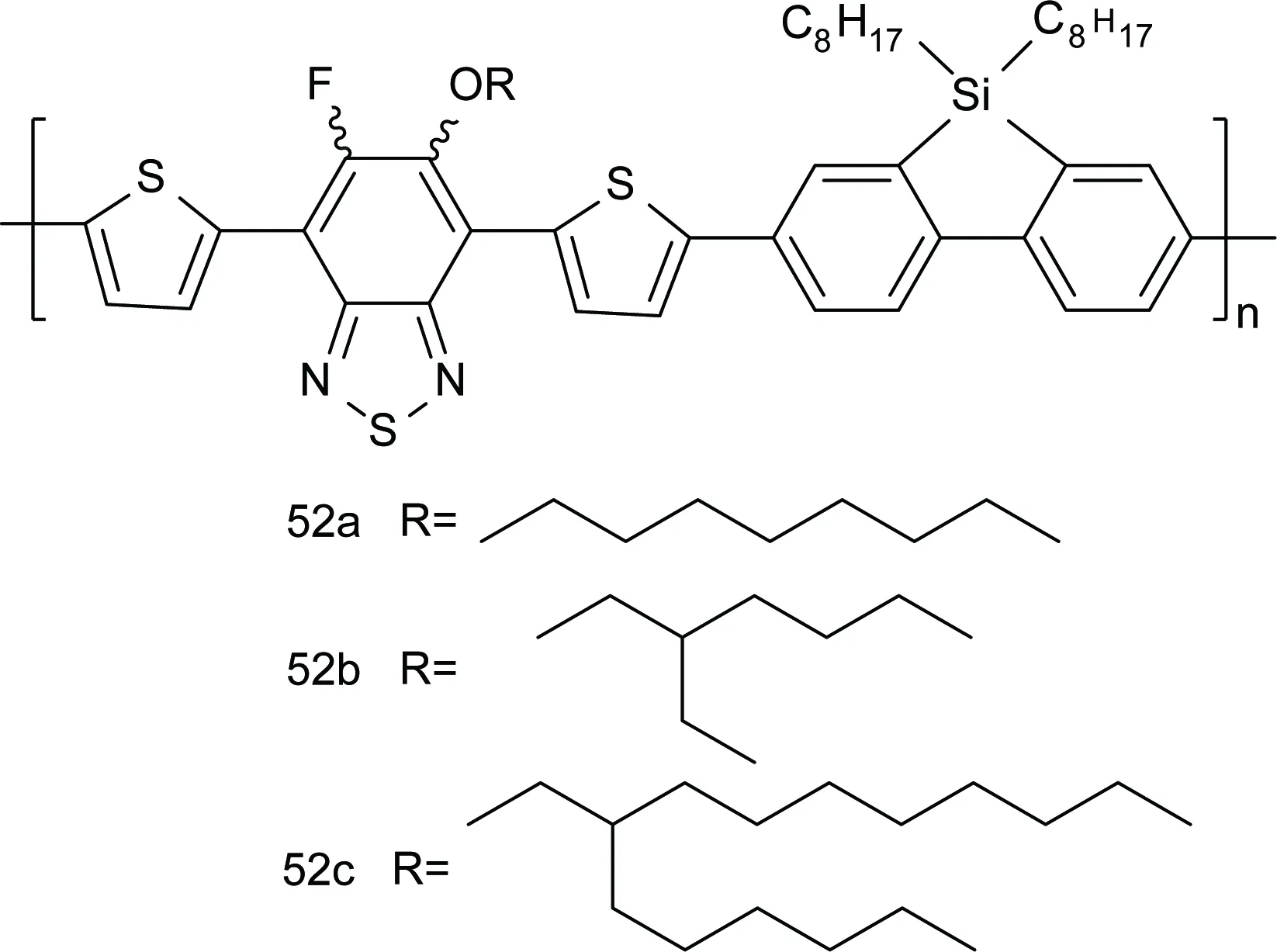
圖12 作為聚合物太陽能電池給體材料的聚合物結構示意圖[58]Fig.12 Structure of the polymers as donor materials of polymer solar cell[58]
Ohshita等[59]最近把DTC、DTS、DTG分別自聚得到3種均聚物(PDTC、PDTS、PDTG),并測試了這3種均聚物的半導體性能,發現PDTS和PDTG具有典型的P型半導體性質和類似的載流子遷移率,而PDTC的載流子遷移率則較低(如圖13)。

圖13 Stille反應合成聚合物PDTS[59]Fig.13 Synthesis of PDTS by Stille reaction [59]
3.3 發光材料
大多數有機發光材料在固態時會呈現發光效率下降甚至不發光的現象,即聚集淬滅現象,極大地限制了其應用范圍,成為有機固體發光材料研究領域的難題。Lu等[60]將Si元素引入到三苯基苯等共軛結構中,制成超支化有機硅聚合物,這類超支化有機硅聚合物顯示出良好的聚集誘導發光效應。
3.4 金屬離子識別
Chen等[14]發現了圖14所示結構能很好地進行光引發的電子轉移,在不同的溶劑中速度為10.8~32.2 ns-1。
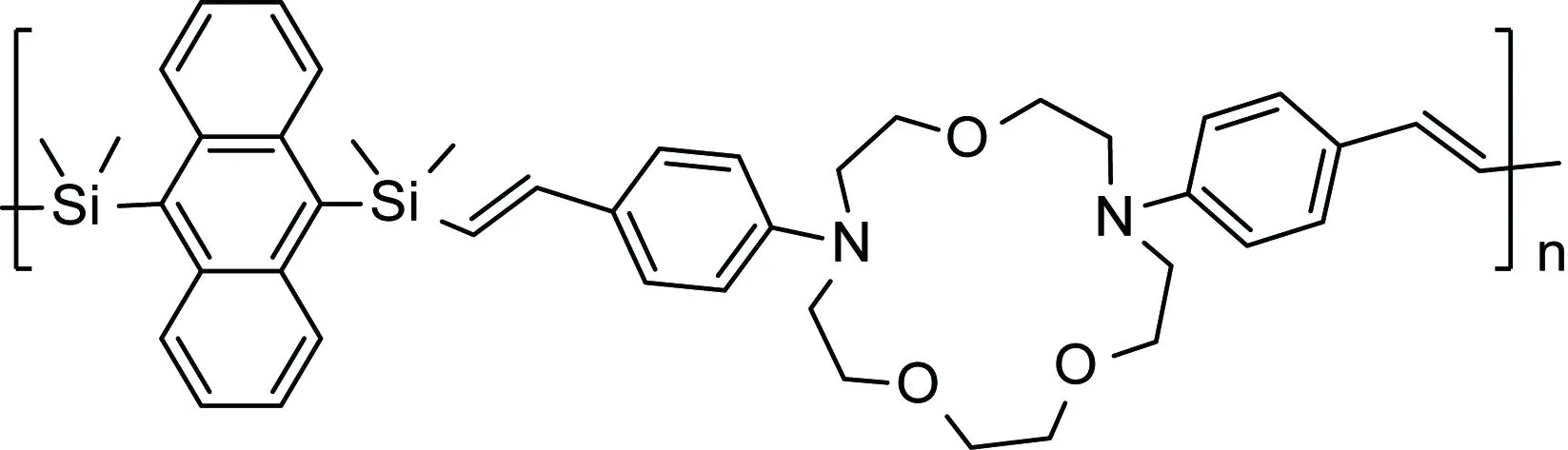
圖14 蒽作為發色團、含氮冠醚為識別單元的聚合物的結構[14]Fig.14 The structure of the polymer containning anthracene as the fluorophore and aminostyrene as the quencher [14]
這類分子能對特定金屬離子進行一定的識別作用,如隨著Pd2+的加入,該化合物的溶液的熒光逐漸增強,可以對金屬離子進行探測。
3.5 爆炸物探測
Sanchez等[61]合成了如圖15所示的化合物,它們能對一些爆炸的化合物進行探測,如TNT、DNT、PA、RDX、HMX、Tetryl、TNG和PETN,探測的量可以達到pg·cm-2級別。
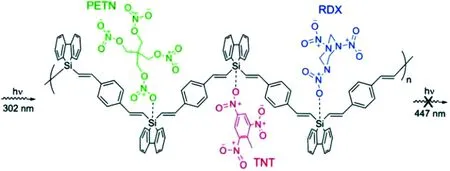
圖15 聚合物對爆炸物檢測的機理示意圖[61]Fig.15 Schematic representation of explosive analyte binding events with polymers [61]
4 結 語
近年來,不飽和聚碳硅烷由于其廣泛的潛在應用獲得了不同領域研究者的關注。不同于傳統的由Si-C-Si鍵構成的聚碳硅烷材料,不飽和聚碳硅烷主鏈由共軛單元與硅共聚而成,硅上烷基的存在有利于增加聚合物的溶解度,且由于Si-C鍵比C-C鍵長,又增加了聚合物的柔性,共軛集團的存在又賦予了聚合物特殊的光物理性能,使得其不僅可以作為陶瓷前驅體,還可用作光致刻蝕劑、電導和光電材料、非線性光學材料以及有機電致發光器件的材料等。設計和開發新型的不飽和聚碳硅烷材料,并拓展其應用領域,如超支化聚合物有希望被應用到生物醫藥領域作為藥物載體和控制藥物釋放。還有把金屬引入到聚合物中,增加陶瓷的產率和更高的熱分解性。不飽和聚碳硅烷的這些性能對各方面的應用都具有潛在的價值,也是未來的研究熱點。
參考文獻 References
[1] Birot M,Pillot J P,Dunogues J.ChemicalReviews[J],1995,95(5):1443-1477.
[2] Yajima S,Hasegawa Y,Hayashi J,etal.JournalofMaterialsScience[J], 1978, 13(12):2569-2576.
[3] Song Maili (宋麥麗),Fu Likun (傅利坤).MaterialsChina(中國材料進展) [J], 2013, 32(04):243-248.
[4] Wang Zhiying (王志英). China, 10604498.9[P]. 2016-10-26.
[5] Zhang Xianbing (張先兵). China,10654386.8[P]. 2017-03-29.
[6] Wang Hu (王 虎). China, 10718858.6[P]. 2017-09-22.
[7] Wang Weigen (王維根). China, 10675137.8[P]. 2017-5-31.
[8] Xu Shengjie (余盛杰). China, 10736516.9[P]. 2017-11-21.
[9] Hu Wenlong(胡文龍). China, 10709987.7[P]. 2017-8-29.
[10] Fang M C,Akira Watanabe A,Matsuda M.Macromolecules[J], 1996, 29(21):6807-6813.
[11] Cao Shiyi(曹適意),Wang Jun(王 軍), Wang Hao(王 浩).ChemicalIndustryandEngineeringProgress(化工進展)[J], 2012, 31(8):1745-1750.
[12] Ohshita J, Kangai S, Yoshida H,etal.JournalofOrganometallicChemistry[J], 2007, 692(4):801-805.
[13] Sunaga T,Ishii J,Yanagibori S,etal. European,1553119[P]. 2005-7-13.
[14] Wang H W,Cheng Y J,Chen C H,etal.Macromolecules[J], 2007, 40(8):2666-2671.
[15] Hua Xiufang(華俢芳),Li Chuanyang(李川陽),Cui Dongmei(崔冬梅).PolymerBulletin(高分子通報)[J],2016, (9):203-226.
[16] Paik K L, Baek N S, Kim H K,etal.Macromolecules[J],2002, 35(18):6782-6791.
[17] Duan X B,Cai G P,Weber W P,etal.ChineseChemicalLetters[J], 2005, 16(3):405-408.
[18] Bai Ying(白 贏),Peng Jiajian(彭家建),Li Jiayun(厲嘉云).ProgressinChemistry(化學進展)[J], 2011, 23(12):2466-2477.
[19] Buisine O, Berthon-Gelloz G, Brière J F,etal.ChemicalCommunications[J], 2005, 36(50):3856-3858.
[20] Truscott B J, Slawin A M Z, Nolan S P.DaltonTransactions[J],2012, 42(1):270-276.
[21] Maifeld S V,Tran M N,Lee D.TetrahedronLetters[J], 2005, 46(1):105-108.
[22] Ba L T, Pink M, Mindiola D J.Organometallics[J, ]2009, 28(7):2234-2243.
[23] Han J W, Hayashi T.TetrahedronAsymmetry[J], 2010, 21(18):2193-2197.
[24] Chalk A J, Harrod J F.JournaloftheAmericanChemicalSociety[J], 1965, 87(1):583-97.
[25] Ojima I, Clos N, Donovan R J,etal.Organometallics[J]1990, 9(12):3127- 3133.
[26] Pang Y, Ijadimaghsoodi S, Barton T J.Macromolecules[J],1993, 26(21):5671-5675.
[27] Xiao Y, Wong R A, Son D Y.Macromolecules[J], 2000, 33(20):7232-7234.
[28] Kim D S, Shim S C.JournalofPolymerSciencePartA:PolymerChemistry[J], 2015, 37(13):2263-2273.
[29] Wang F,Kaafarani B R,Neckers D C.Macromolecules[J],2003,36(22):8225-8230.
[30] Kim D S,Shim S C.JournalofPolymerSciencePartA:PolymerChemistry[J],1999,37(15):2933-2940.
[31] Yamashita H,Leon M S D,Channasanon S,etal.Polymer[J],2003, 44(23):7089-7093.
[32] Yamashita H,Suzuki Y,Rao T V.JournalofOrganometallicChemistry[J],2012,710(7):59-67.
[33] Mori A,Takahisa E,Kajiro H.ChemistryLetters[J],1998,27(5):443-444.
[34] Mori A,Takahisa E,Kajiro H,etal.Macromolecules[J],2000,33(33):1115-1116.
[35]Mori A,Takahisa E,Yamamura Y,etal.Organometallics[J],2004,23(8):1755-1765.
[36] Kwak G,Sumiya K I,Sanda F,etal.JournalofPolymerSciencePartA:PolymerChemistry[J],2003,41(22): 3615-3624.
[37] Sumiya K I,Kwak G,Sanda F,etal.JournalofPolymerSciencePartA:PolymerChemistry[J],2004,42(11):2774-2783.
[38] Chen R M,Chien K M,Wong K T,etal.JournaloftheAmericanChemicalSociety[J],1997,119(46):11321-11322.
[39] Chen R M,Luh T Y.Tetrahedron[J],1998,54(7):1197-1206.
[40] Cheng Y J,Luh T Y.ChemistryAEuropeanJournal[J],2004,10(21):5361-5368.
[41]Perry R J,Karageorgis M,Hensler J.Macromolecules[J],2007,40(11):3929-3938.
[42] Sasano T,Sogawa H,Tamura K,etal.JournalofPolymerSciencePartA:PolymerChemistry[J],2010,48(8):1815-1821.
[43] Zhao Z,Jiang T,Guo Y,etal.JournalofPolymerSciencePartA:PolymerChemistry[J],2012,50(11):2265-2274.
[44] Zhao Z,Guo Y,Jiang T,etal.MacromolecularRapidCommunications[J],2012,33(12):1074-1079.
[45] Schmidt M,Durif C,Acosta E D,etal.ChemistryAEuropeanJournal[J],2017,23(67):17103-17117.
[46] Ji P,Pei X,Miao Y,etal.AdvancesinAppliedCeramics[J],2017,116(8):445-451.
[47] Zhong X,Pei X,Miao Y,etal.JournaloftheEuropeanCeramicSociety[J],2017,37(10):3263-3270.
[48] Ma Z,Xiao C,Liu C,etal.OrganicLetters[J],2017, 19(16):4331-4334.
[49] Kim H K,Ryu M K,Lee S M.Macromolecules[J],1997,30(4):1236-1239.
[50] Tominaga Y,Nanthana V,Tohyama D.PolymerJournal[J],2012,44(12):1155-1158.
[51] Nakamura M,Tominaga Y.ElectrochimicaActa[J],2011,57(1):36-39.
[52] Nanthana V,Tominaga Y.KōbunshiRombunShū[J],2013,70(1):23-28.
[53] Tominaga Y,Shimomura T,Nakamura M.Polymer[J],2010,51(19):4295-4298.
[54] Mindemark J,Imholt L,Brandell D.ElectrochimicaActa[J],2015,175(2015):247-253.
[55] Mindemark J,Imholt L,Montero J,etal.JournalofPolymerSciencePartA:PolymerChemistry[J],2016,54(14):2128-2135.
[56] Matsumoto K,Endo T,Katsuda K,etal.JournalofPolymerSciencePartA:PolymerChemistry[J],2012,50(24):5161-5169.
[57] Matsumoto K,Kakehashi M,Ouchi H,etal.Macromolecules[J],2016,49(24):9441-9448.
[58] Li G W,Kang C,Gong X,etal.Macromolecules[J],2014,47(14):4645-4652.
[59] Ohshita J,Miyazaki M,Tanaka D,etal.PolymerChemistry[J],2013,4(10):3116-3122.
[60] Lu P,Lam J W,Liu J,etal.MacromolecularRapidCommunications[J],2010,31(9-10):834-839.
[61] Sanchez J C,Urbas S A,Toal S J,etal.Macromolecules[J],2008,41(4):1237-1245.
