葛根素標準樣品的研制
2018-04-19 03:40:36
分析儀器 2018年2期
(1.北京市理化分析測試中心,有機材料檢測技術與質量評價北京市重點實驗室,北京市科學技術研究院分析測試技術重點實驗室,北京 100089;2.中國標準化研究院,北京 100191)
葛為多年生豆科纏繞藤本植物,側蔓和須根多,但塊根深生,呈紡錘形或長棒形,表皮為淡黃色,有皺褶,是主要食用和藥用部分。葛根為野葛(Pueraria Lobata(Willd.) Ohwi)或粉葛(P. thomsomi Benth)的干燥根,含有異黃酮類、三萜類、芳香類等活性成分。其中異黃酮類化合物是葛根的主要化學成分。葛根味甘、辛,性涼,歸肺、胃經,具有解肌退熱、透疹、生津止渴、升陽止瀉等功效[1],被廣泛用于保護心腦血管[2-4]、治療感冒[5]、Ⅱ型糖尿病[6]、腹瀉等中藥復方制劑中,用量極大,是制劑的重要成分。其中葛根素是葛屬植物的特有成分,亦是主要有效成分,具有擴張冠狀動脈血管,降低血脂,增強心肌收縮力,降低血壓,防治高血壓頭暈、頭痛、頸項疼痛[7-12]等功效。
市場上葛根相關的產品種類繁多,其中葛根素作為葛根主要的化學成分,其含量的檢測需要該標準樣品作為檢測對照;同時,在葛根相關活性研究和產品開發中,葛根素也是重要的研究對象,因此迫切需要開展該標準樣品的研制工作。本研究采用高速逆流色譜(high-speed countercurrent chromatography,HSCCC)技術制備葛根素,通過紫外吸收光譜(ultraviolet absorption spectrometry,UV)、紅外光譜(infrared spectroscopy,IR)、質譜(mass spectrometry,MS)和核磁共振(nuclear magnetic resonance,NMR)波譜等方法進行結構鑒定,并開展均勻性檢驗、穩定性檢驗、定值研究,最終得到純度大于98%、擴展不確定度小于1%的葛根素標準樣品。
1 材料與方法
1.1 儀器與材料
KQ-250E超聲波清洗器(昆山市超聲儀器有限公司);TBE-300B半制備型高速逆流色譜儀(上海同田生物技術有限公司);LC-20高效液相色譜儀(日本Shimadzu公司);RE-2000旋轉蒸發儀(上海亞榮生化儀器廠);ALPHR1-2冷凍干燥機(德國Marin Christ公司);Spectrum400傅立葉變換紅外-近紅外光譜儀(美國PerkinElmer公司);1100 Series LC/MSD SL(美國Agilent公司),超導核磁共振譜儀(德國Bruker公司)。
乙酸乙酯,甲醇,正丁醇(分析純,國藥集團化學試劑有限公司);乙腈(色譜純,美國Fisher Scientific公司);葛根提取物(湖南長沙遠航生物制品有限公司)。
1.2 方法
1.2.1 葛根素樣品制備與分裝
HSCCC溶劑體系為乙酸乙酯∶正丁醇∶水(2∶1∶3,v/v),轉速900 r/min,流速1.5 mL/min,分離溫度25℃,檢測波長254 nm。根據HSCCC圖譜收集目標化合物,旋轉蒸發去除溶劑,冷凍干燥后得到高純度葛根素樣品。
將葛根素樣品采用2 mL棕色樣品瓶進行分裝。分裝是在相對獨立和潔凈空間進行的,以每瓶10 mg分裝,用十萬分之一天平稱量,樣品共100 瓶,以1~100號計。分裝好的樣品瓶放在4℃冰箱中長期保存。
1.2.2 純度分析
配制濃度為0.5 mg/mL的葛根素樣品溶液,采用HPLC峰面積歸一法對葛根素進行純度分析。色譜柱為Wondasil C18(5 μm, 150×4.6 mm) ;流動相∶乙腈∶水=12∶88;流速:1.0 mL/min;柱溫:30℃;檢測波長:254 nm;運行時間:30 min。
1.2.3 結構確證
采用UV、IR、MS、NMR進行結構確證。UV分析條件:甲醇作為溶劑,濃度為0.1 mg/mL,掃描范圍200~400 nm;IR分析條件:KBr壓片法,掃描范圍400~4000 cm-1。MS分析條件:正、負兩種離子模式,錐孔氣流速40 L/min,毛細管電壓3.0 kV,脫溶劑溫度320℃,質量掃描范圍m/z150~1000。NMR分析條件:溶劑為氘代丙酮加重水,采集13C-NMR和1H-NMR譜圖。
1.2.4 均勻性、穩定性、定值研究
依據GB/T 15000.3—2008《標準樣品工作導則(3)標準樣品:定值的一般原則和統計方法》[13]開展均勻性檢驗、穩定性檢驗、定值研究工作。均勻性:采用方差分析法進行純度均勻性檢驗,判斷葛根素的均勻性是否合格。穩定性:將葛根素樣品避光貯存于4℃,開展12個月長期穩定性的研究。采用直線作為經驗模型,觀察斜率值是否有顯著變化,預測葛根素的穩定性變化。定值:采用多個實驗室協作試驗的聯合定值方式,分別對隨機抽取8個樣品進行檢測,對所采集的測定結果采用格拉布斯(Grubbs)檢驗法進行檢驗,計算葛根素標準樣品的標準值和不確定度。
2 結果與分析
2.1 葛根素制備及純度分析
采用HSCCC乙酸乙酯∶正丁醇∶水(2∶1∶3,v/v)溶劑體系分離純化得到高純度葛根素樣品(圖1),采用HPLC對葛根提取物和分離所得單體進行洗脫,254 nm檢測波長下,應用峰面積歸一法計算純度值,分離所得單體葛根素的純度為99.53%。(圖2)

圖1 葛根素HSCCC圖

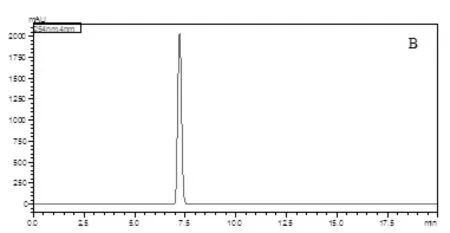
圖2 葛根素HPLC圖A.葛根提取物;B.分離所得單體
2.2 結構確證
采用UV、 IR、 MS、NMR對葛根素進行結構鑒定。UV最大吸收波長為250 nm(圖3); IR吸收峰(圖4):3240(-O-H),1634(-C=O),1515,1446(Ar),1260,1212,1178,1082(醚-C-O)。通過ESI-MS正負離子模式掃描(圖5)可得:[M+Na]+=439.2,[M-H]-=415.1,故測定的相對分子質量為416。1H-NMR和13C-NMR測試結果見圖6,通過對數據進行歸屬(表1、表2)。與文獻[14,15]進行比較,確定該樣品為葛根素。
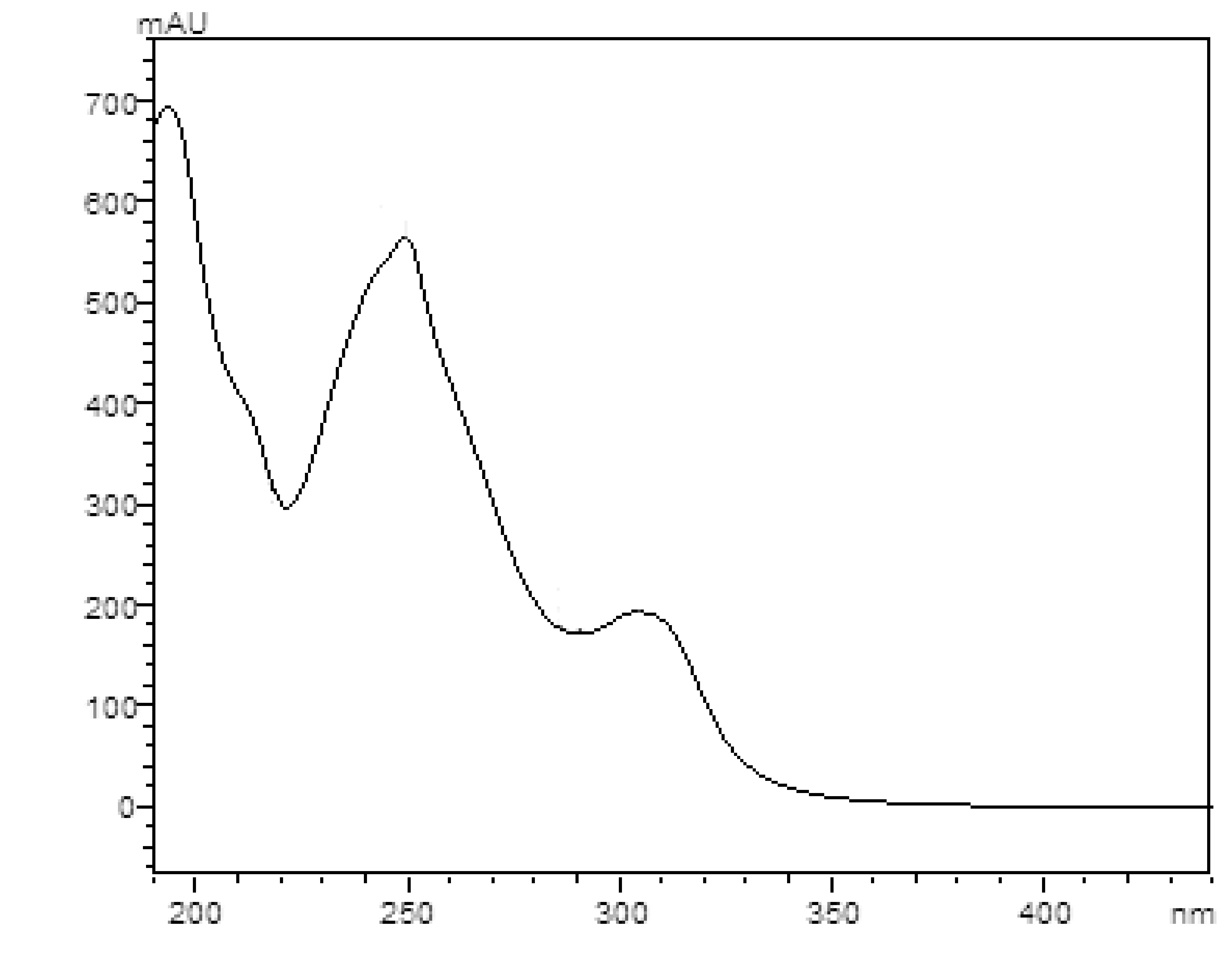
圖3 葛根素紫外吸收光譜圖
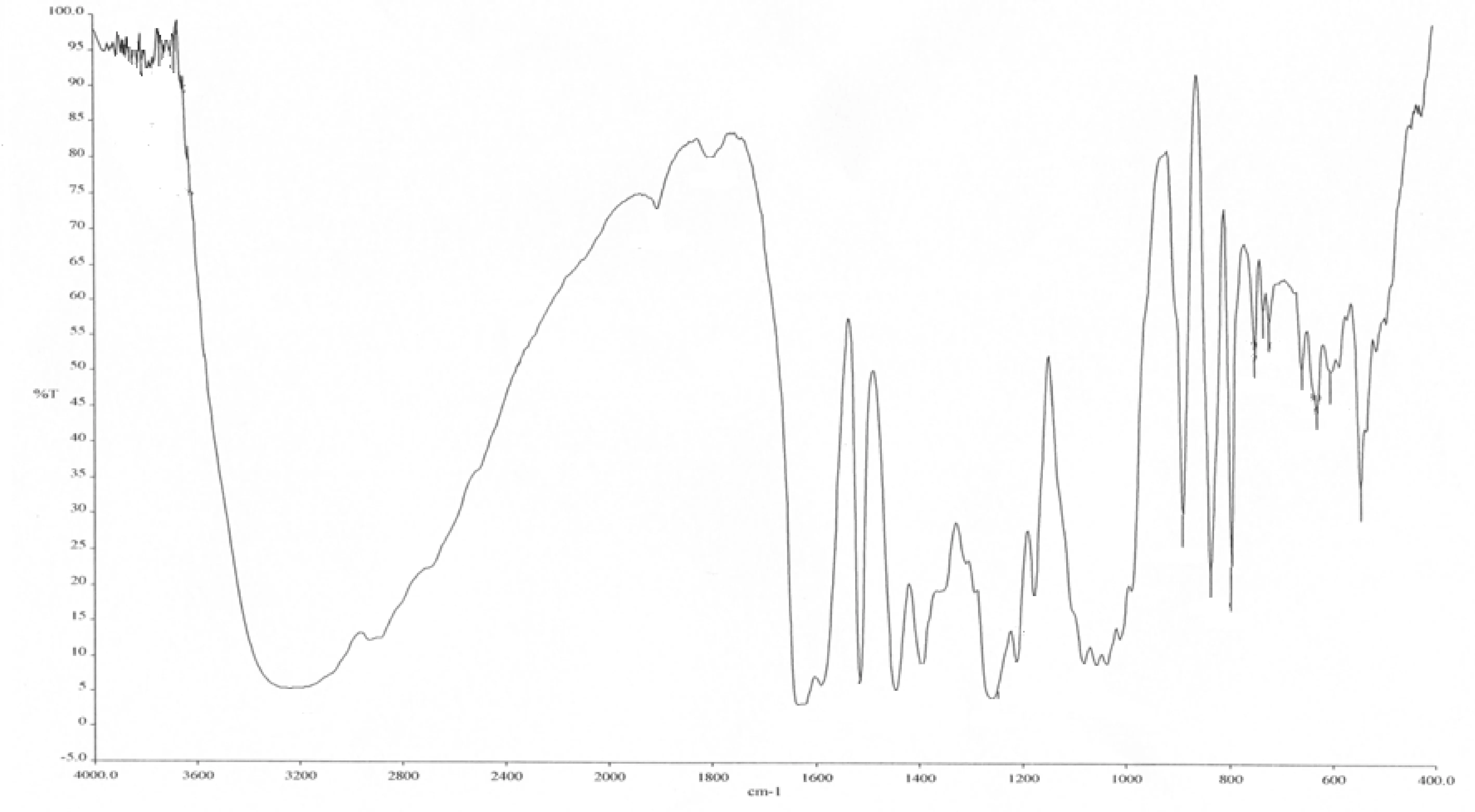
圖4 葛根素紅外光譜圖
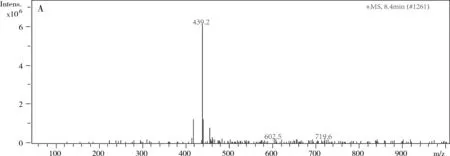
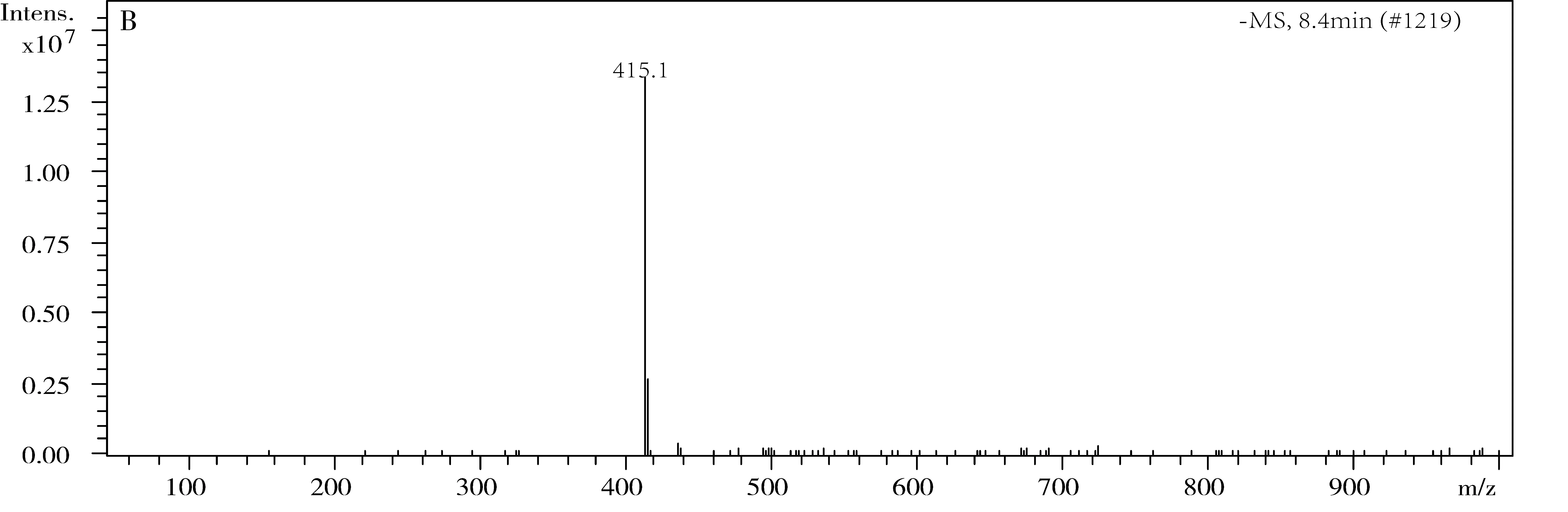
圖5 葛根素質譜圖A.正離子模式;B.負離子模式下
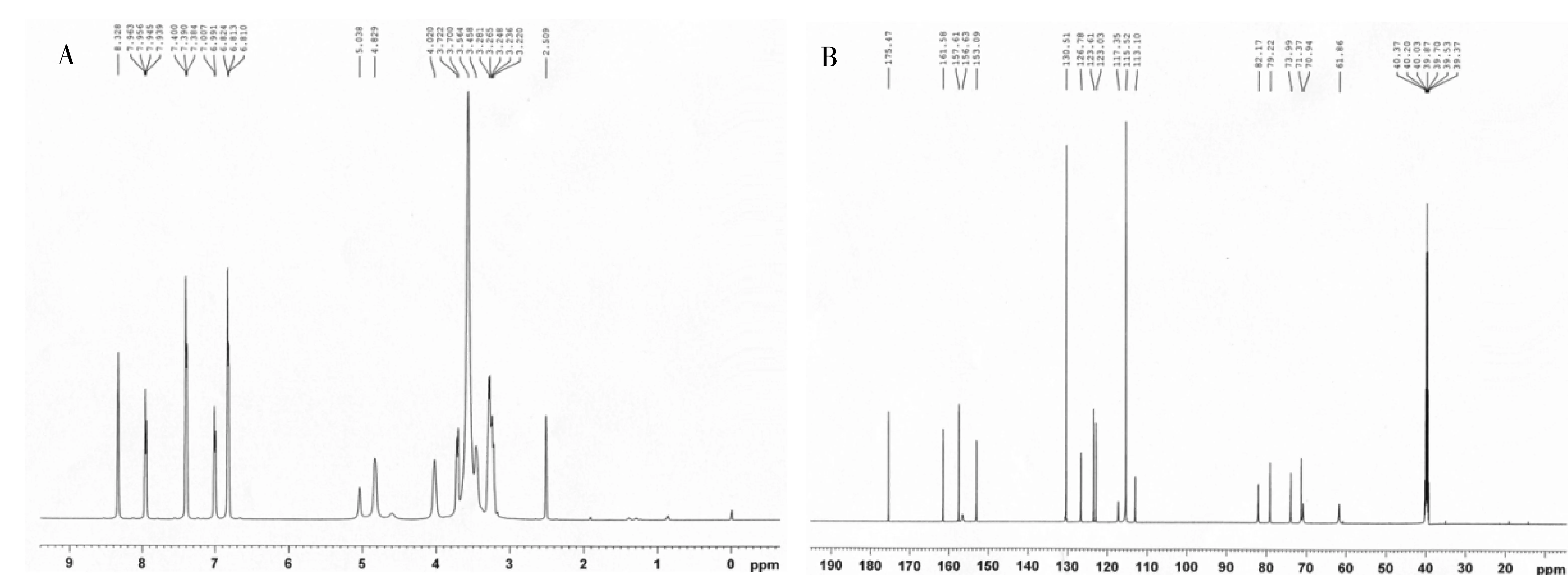
圖6 葛根素核磁共振圖譜A.1H-NMR;B. 13C-NMR
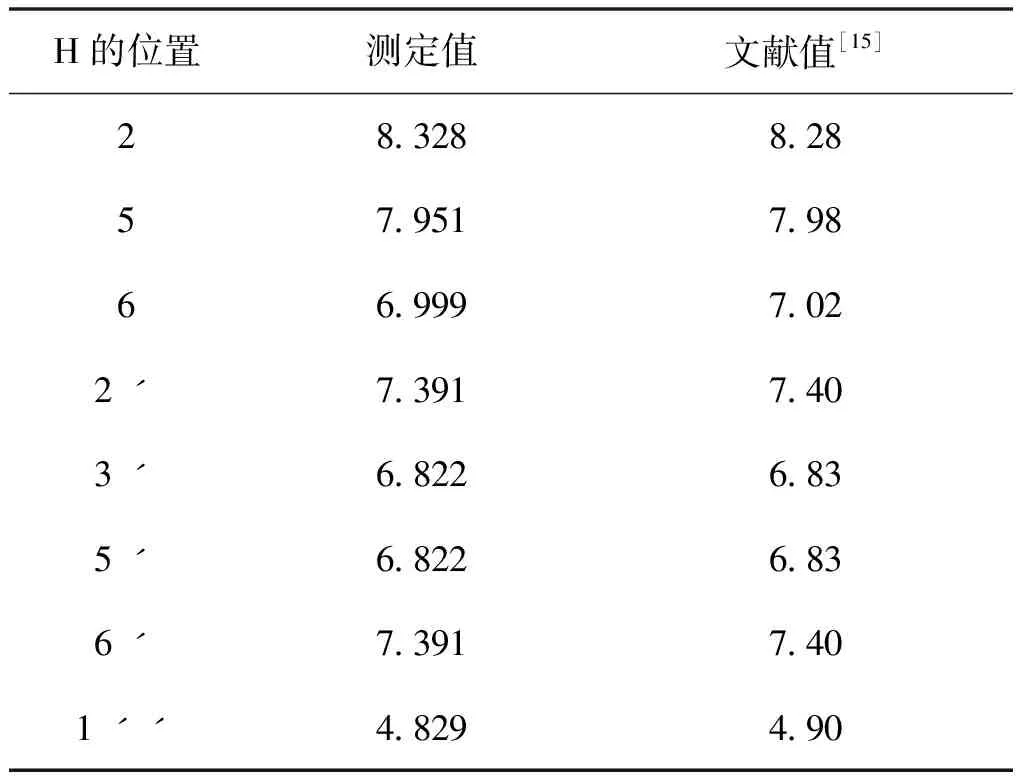
H的位置測定值文獻值[15]2832882857951798669997022ˊ73917403ˊ68226835ˊ68226836ˊ73917401ˊˊ4829490
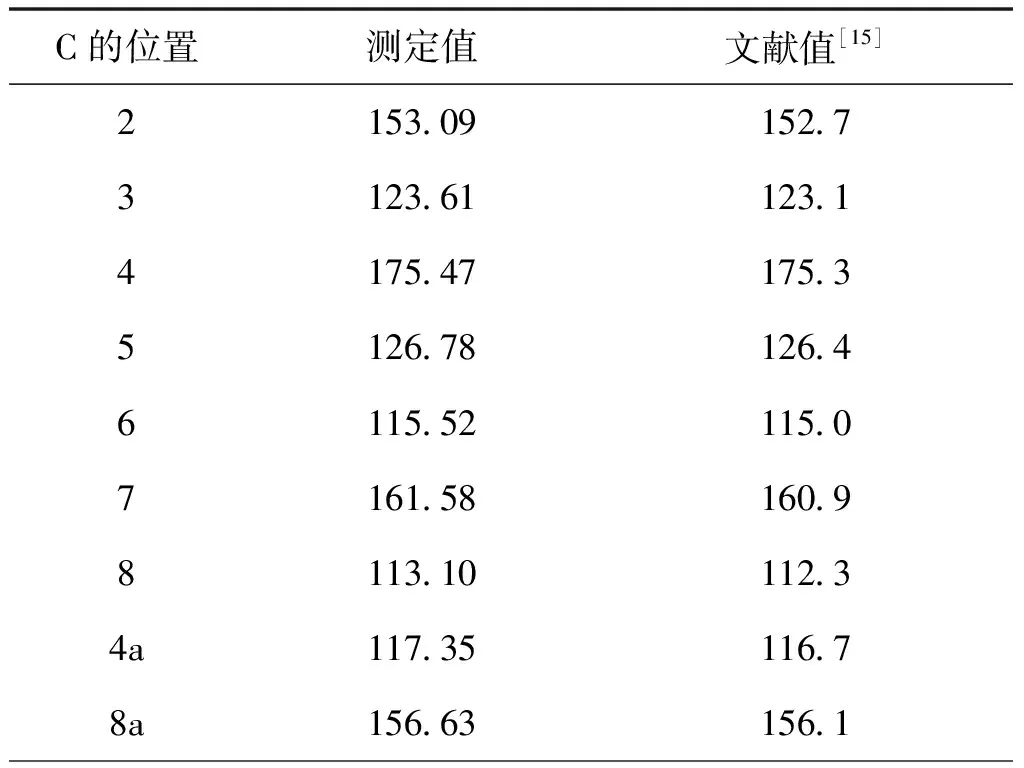
表2 13C-NMR測試數據
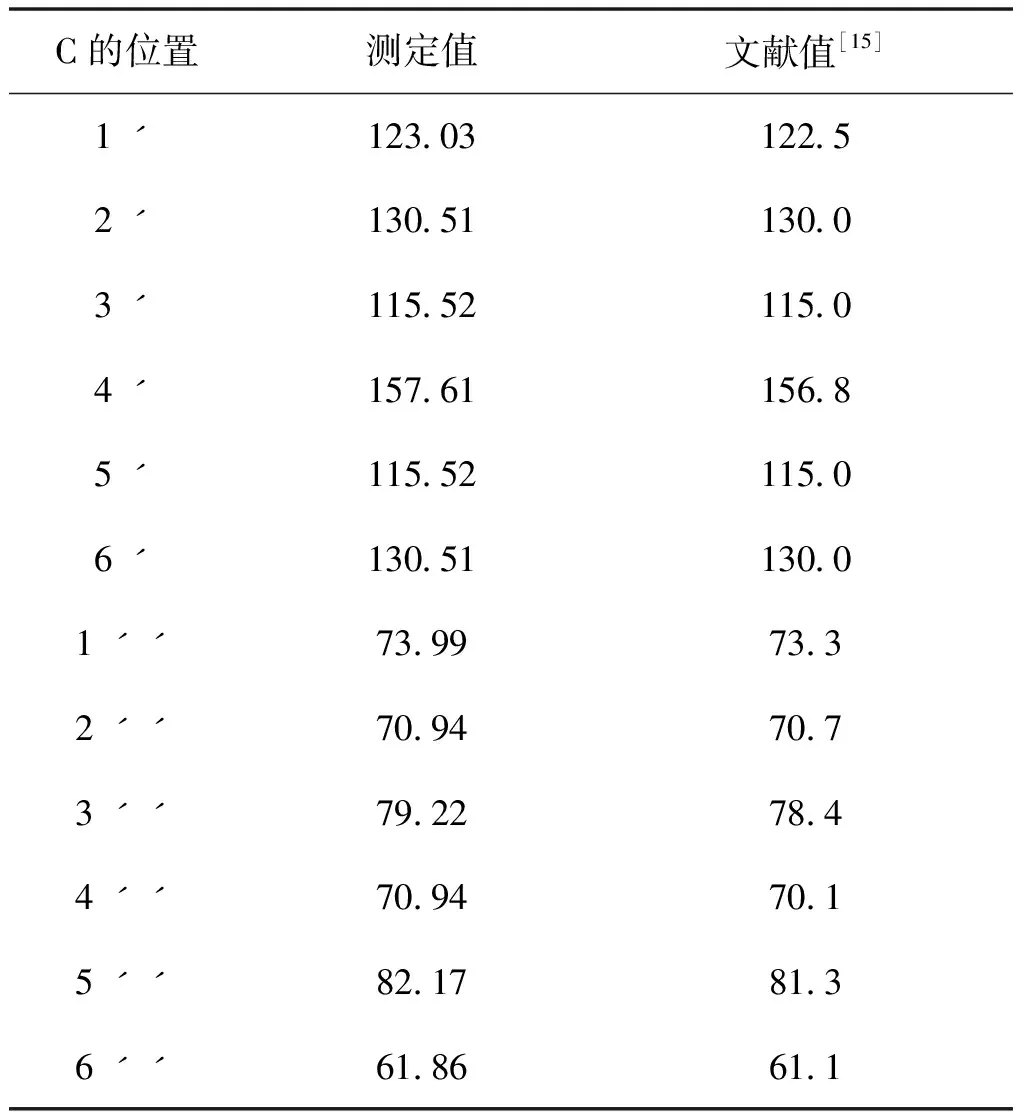
續表2
2.3 均勻性檢驗
由于所制備的樣品單元數都小于100,根據隨機數表,分別抽取10瓶樣品進行均勻性檢驗。其中葛根素瓶號為42、9、67、89、53、63、32、75、36、60;每瓶樣品各稱3份,每份1.0 mg按照1.2.2純度分析方法進行HPLC分析,計算其純度。采用方差分析法進行檢驗,對其均勻性做出判斷。結果見表3。

表3 根素樣品均勻性檢驗結果

2.4 穩定性檢驗
將葛根素樣品于4℃冰箱冷藏避光保存,以1年為期,對制備的葛根素樣品每3個月取樣,按1.2.2純度分析方法,峰面積歸一化法進行純度計算,每份試樣平行測試3次,采用t檢驗對數據進行分析,穩定性檢驗結果見表4。
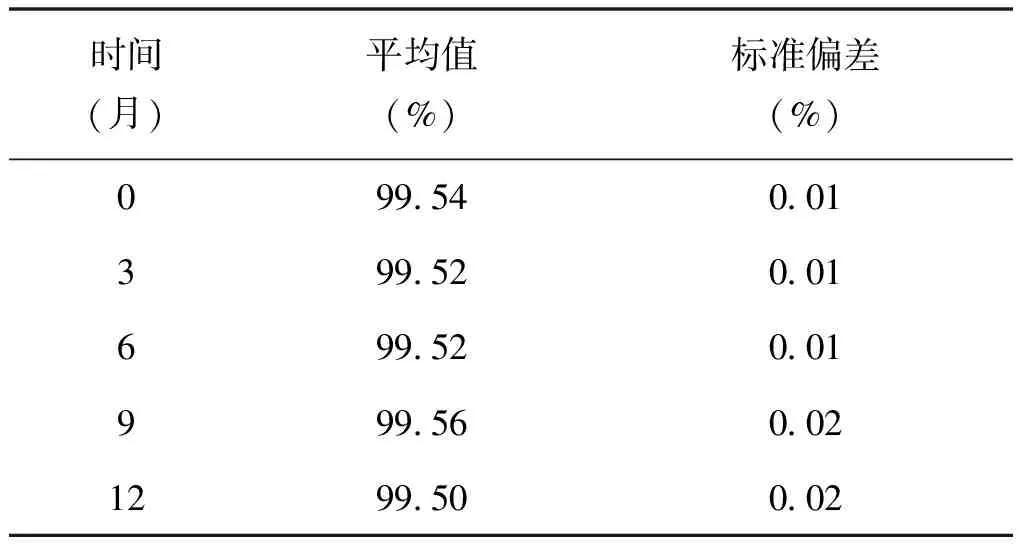
表4 穩定性檢驗結果
采用直線作為經驗模型,觀察斜率值是否有顯著變化,以此對標準樣品的穩定性變化進行預測。斜率可用下式計算:
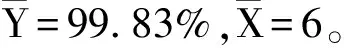
直線上的點的標準偏差可由下式計算:
式中穩定性測量次數n=3。
取其平方根s=0.276%,與斜率相關的不確定度用下式計算:
自由度為n-2和p=0.95(95%置信水平)的分布t因子等于3.128,由于|b1| 對聯合定值的8家實驗室所采集的測定結果,采用Grubbs檢驗法進行檢驗,未發現異常值。經過數據統計,計算得出葛根素樣品的標準值和不確定度。不同實驗室檢測和方差分析結果見表5、表6。 表5 不同實驗室檢測結果 表6 不同實驗室方差分析結果 總平均值由下式計算: 總平均值的不確定度等于: =7.43×10-5, 式中,實驗室內測量次數n=6,實驗室數量p=8, 在置信概率為95% 時,k=2,則擴展不確定度UCRM=2uCRM=0.70%。 針對我國尚無葛根素國家實物標準樣品的現狀,開展了葛根素標準樣品的研制工作,成功的建立了葛根素樣品的制備方法。并依據GB/T 15000標準樣品工作導則,對制備得到的葛根素樣品進行均勻性檢驗、穩定性檢驗及定值研究,最終申報獲批成為國家實物標準樣品。 葛根素標準樣品的標準值為99.51%,擴展不確定度(95%置信區間)為0.70%。該標準樣品符合GB/T 15000標準樣品工作導則要求,填補了國內該領域研究的空白。葛根素標準樣品不僅可以滿足葛根及相關產品分析檢測、質量控制工作的需求,同時,也為檢測結果的準確性、可比性,以及溯源性提供了技術支撐。 [1]張曉娟,周海純.葛根化學成分,現代藥理及臨床應用研究進展[J].中醫藥信息,2017, 34(1):124-126. [2]張軍霞.使用葛根素治療腦梗死的效果分析[J].當代醫藥論叢,2017,15(2):143-144. [3]何素珍.葛根素注射液聯合丁苯肽治療腦梗死的臨床價值[J].世界臨床醫學,2017,11(1):94. [4]郭娜,焦黎明,閆冬雪.頭針結合天麻葛根顆粒治療后循環缺血性眩暈的臨床療效觀察[J].中西醫結合心腦血管病雜志,2016,14(23):2741-2743. [5]趙益,賴小東,葉爭榮,等.葛根芩連湯對潰瘍性結腸炎模型大鼠抗氧化及抗炎的作用機制 [J].中華中醫藥雜志,2016, 34(5):1741-1745. [6]王蘭,藍璟,龔頻,等.葛根異黃酮降血糖活性及作用機制的研究[J].食品科技,2017(3):223-226. . [7]國醫學科學院藥物研究所. 葛根的臨床應用和實驗研究[J]. 醫藥研究通訊. 1972, (2): 14. [8]Koichi T, Hideji I. Isoflavonoids and the other constituents in callustissues of pueraria lobala[J]. Chemical and Pharmaceutical Bulletin, 1982, 30(4): 1496. [9]鐘平華. 葛根素的現代應用概況[J]. 中國食品衛生雜志. 1997, 9(6): 36-37. [10]He X L, Tan T W, Xu B Z et a1. Separation and purification of puerafin using 13-cyclodextrin-coupled agarose gel media[J]. Journal of Chromatography A, 2004, 1022: 77-82. [11]李為厚, 武維恒.葛根素注射液治療冠心病心絞痛的臨床研究[J].華北煤炭醫學院學報. 2000, 2(6): 611-612. [12]羅偉, 李保東, 楊瑞華, 等.葛根素治療高血壓病的臨床研究[J].中國中醫基礎醫學雜志. 2000, 6(5): 61-63. [13] GB/T15000.3-2008,標準樣品工作導則(3)標準樣品定值的一般原則和統計方法[S],2008. [14]雷雨,圖雅,張艷玲,等.野葛與粉葛的二維紅外相關光譜鑒別[J].分析儀器,2010,(3):47-49. [15]Kinji J E, Furusawa J I, Baba J, et al. Studies on the constituents of Pueraria lobata. III Isoflavonoids and related compounds in the roots and the voluble stems[J]. Chemical and Pharmaceutical Bulletin,1987,35: 4846-4850.2.5 定值



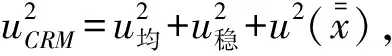
3 結論
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
