伴強直發作的Dravet綜合征2例報告并文獻復習
2018-03-30 01:43:59魏春苗夏桂枝任榕娜
東南國防醫藥 2018年2期
魏春苗,夏桂枝,任榕娜
0 引 言
Dravet綜合征(Dravet syndrome,DS)是一種少見的嬰兒期起病的難治性早發性癲癇腦病。發病率為1/22 000~1/40 000[1-2],死亡率高且有癲癇猝死的風險[3-4]。一方面DS患兒發病前生長發育正常,1歲以內以熱性驚厥起病,早期神經影像學及腦電圖無異常發現,診斷困難;另一方面DS患兒對抗癲癇藥物療效差,1歲以后多出現精神運動發育落后或倒退且癥狀可持續到成年;因此早期診斷、合理治療尤為重要。近年來SCN1A基因突變的發現為DS的早期診斷提供了重要依據,對合理用藥也具有一定的指導作用。現將2例伴強直發作的DS少見表型患兒的臨床及基因突變特點進行分析,結合文獻復習報道如下。
1 資料與方法
1.1一般資料收集2011年1月至2017年3月在福州總醫院兒科神經專科門診及病房診治的2例伴強直發作的DS患兒的臨床資料,包括發病年齡、性別、臨床表現、個人史、家族史、實驗室檢查、視頻腦電圖,頭顱MRI、診療過程及隨訪情況。
病例1:7歲女性患兒,足月順產,父母之間無血緣關系,無癲癇及熱性驚厥家族史。5月齡時首次于體溫38 ℃左右出現驚厥發作,表現為意識喪失,四肢陣攣性抽動,持續約10~15 min自行緩解。此后反復出現發熱或感染后類似發作,2~3歲發作頻繁,3歲以后發作次數減少,約3個月發作1次。 4歲時出現3次不典型失神發作。5歲后出現反復強直發作,表現為意識喪失,雙眼上翻,四肢強直,頭后仰,持續5~8 min自行緩解,緩解后全身無力,意識逐漸恢復,發作前有熱或無熱,每年2~3次。曾先后給予托吡酯、左乙拉西坦、丙戊酸鈉、奧卡西平、氯硝西泮等藥物治療,發作控制不佳,現用丙戊酸鈉聯合托吡酯治療。首次發作及發作1年后2次查頭顱MRI及視頻腦電圖(vedio electroencephalogram,VEEG)未見異常。6歲時VEEG:①基本波率為5~7次/s慢波節律,波形不整,左右波幅不對稱,調節調幅差,兩顳枕區慢波節律優勢,時而高波幅同步陣發;②異常腦波:中-高波幅同步陣發慢波節律,兩側波幅不對稱,兩額顳、(旁)中線區夾雜尖棘慢波。血電解質、血氨、血漿乳酸、血尿遺傳代謝病篩查均未見異常。8個月時Gesell發育診斷量表評估:99;6歲韋氏兒童智能測試:85,感覺統合嚴重失調;現上小學,學習成績較差。
病例2:3歲5個月男性患兒,足月順產,父母之間無血緣關系,無癲癇及熱性驚厥家族史。首次于8月齡感冒后發熱,體溫38.5 ℃左右出現右側肢體陣攣性抽動,隨后出現意識喪失、雙眼上翻、牙關緊閉、口唇青紫、四肢強直陣攣,前后持續約15 min自行緩解,次日再次發作1次,以后間隔幾天~1個月發作1次,有時1天數次,1~3歲發作次數稍有減少,間隔2~3個月1次,有熱或無熱,表現同前。3歲后出現2次反復強直發作,表現為意識喪失、雙眼上翻、牙關緊閉、四肢強直呈角弓反張狀,無陣攣出現,約5 min后自行緩解,緩解后全身無力,意識逐漸恢復。10個月查頭顱MRI、VEEG未見異常;2歲時VEEG:①兩側波幅不對稱、調節調幅差,同步陣發4~6 Hz慢波節律,兩后頭部(旁)中線區優勢;②廣泛的尖棘慢波節律,旁中線區優勢。2個月前復查VEEG無明顯變化。血電解質、血氨、血漿乳酸、血尿遺傳代謝病篩查均未見異常。曾先后口服左乙拉西坦、丙戊酸及托吡酯治療,療效不佳,現用丙戊酸聯合托吡酯治療。10個月Gesell發育診斷量表評估:95;2歲Gesell發育診斷量表評估:80;現走路易跌倒;語言發育明顯落后。
1.2方法采集患兒及父母外周血2 mL(EDTA抗凝),用BloodGen Midi Kit (CWBIO, China) 提取患兒全基因組DNA,操作按照試劑盒說明書進行。參考文獻與OMIM數據庫信息,將OMIM數據庫中所有與四千種單基因遺傳病相關的基因組外顯子區域定制羅氏NimbleGen捕獲探針,然后制備捕獲文庫、Illumina平臺進行二代測序,最后進行數據分析。根據所驗證位點序列設計引物,采用PCR方法進行擴增,ABI 3730XL 測序儀進行一代測序驗證,測序引物采用原PCR引物,然后進行基因序列分析和比對。二代測序和一代測序驗證均委托北京智因東方轉化醫學研究中心完成。
2 結 果
2.1臨床特點2例患兒分別在出生5個月和8個月起病,熱性驚厥為其首次發作表現。發作形式中除有常見的全身及偏側陣攣、強直陣攣和不典型失神等多種發作外,都出現了DS少見的強直發作。2例患兒均無癲癇及熱性驚厥家族史,實驗室檢查及頭顱MRI未見異常,視頻腦電圖為DS特征性棘慢波發放。2例患兒先后應用多種抗癲癇藥物,但療效均不佳,起病前發育正常,現都有不同程度的精神運動發育落后。
2.2基因突變特點病例1:四千種單基因病基因檢測結果:SCN1A變異,chr2:166904182,c.1125G>C(E10),P.315.L>F,雜合,其父母均未發現相同突變,為新發錯義突變,見圖1a。病例2:四千種單基因病基因檢測結果:SCN1A變異,chr2: 166894356,c.2876G>C(E17),P.959,C>S,雜合,父母均未發現相同突變,為新發錯義突變,見圖1b。
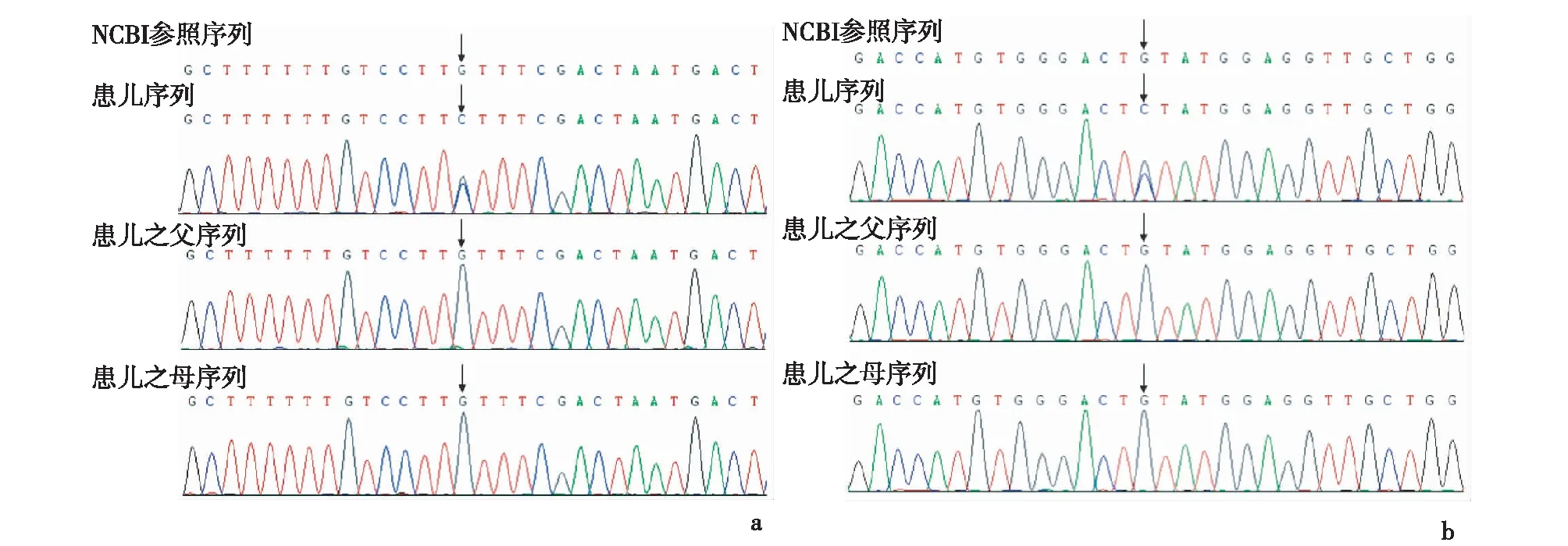
a:病例1(c.1125G>C);b:病例2(c.2876G>C)圖1 Dravet綜合征SCN1A突變一代測序峰圖(箭頭所指為突變位點)
3 討 論
DS的臨床核心表現為:①1歲以內起病,首次發作為持續時間較長的熱性驚厥,或疫苗接種、洗熱水澡等誘發的無熱驚厥;②1歲后逐漸出現多種形式的無熱驚厥,包括全身或半側陣攣、強直陣攣發作、肌陣攣發作、不典型失神、局部性發作等,極少數病例出現強直發作,常發展為癲癇持續狀態;③首次發作時生長發育正常,1歲后逐漸出現精神運動發育落后或倒退,可出現共濟失調、錐體束征及運動不協調;④神經影像學多無異常;⑤腦電圖檢查在1歲以前多無異常,以后逐漸出現θ波為主的慢活動或廣泛性、局灶性的癲癇放電;⑥對抗癲癇藥物治療效果差[5-6]。DS的診斷主要依據上述核心癥狀。
特別的是本報道中2例患兒在病程中均出現了強直發作這一DS少見的發作形式,表現為軸性強直,例1在5歲后出現,例2在3歲后出現,均在癲癇頻繁發作期后,發作次數較前明顯減少。在對DS的首次描述中并未提及強直發作[7],之后表現為強直發作的DS病例陸續被報道,但例數均較少[8-9],最新的文獻對DS核心癥狀的描述中,仍然認為強直發作是不常見的發作形式[10-11]。強直發作表現類似Lennox-Gastaut綜合征(LGS)的軸性強直,有時伴有肌陣攣發作,強直發作往往是散發的,在一次發作過程中反復重復出現。Dravet等[8]報道的發作期腦電圖表現為電活動減少或低波幅的快波節律后出現慢波或間隔以電活動減少的快速的募集節律,發作間期睡眠腦電圖表現為與LGS相同的快波節律和多棘波發放。Nabbout等[9]報道強直發作間期腦電圖為前額區的慢的雙向或三向棘波,后伴或不伴有慢波。本報道中2例患兒未記錄到上述強直發作的腦電圖表現,但臨床表現與報道的DS的強直發作基本相同。
目前對DS研究最大的進展是DS基因突變的發現。2001年Claes等[12]檢測出7例DS患兒存在編碼鈉離子通道α亞單位的SCN1A基因新發突變,隨后的研究發現70%~80%的DS患兒攜帶SCN1A基因突變[9,13],這些突變發生于SCN1A不同的基因位點,90%為新發突變,有截斷突變、錯義突變、無義突變、缺失突變等多種突變形式[14]。雖然到目前為止,基因型和表現型之間的關系尚未闡明,并不是所有攜帶SCN1A基因突變的都是DS,但SCN1A基因突變對于DS尤其是少見表型病例的早期診斷仍是一個非常有力的診斷依據[9]。本報道中2例患兒出現DS少見的強直發作,早期診斷更加困難,最終基因檢測出SCN1A基因新發錯義突變而確診,可見基因檢測對于DS的早期診斷至關重要。近年來在一些臨床表現類似DS而SCN1A基因突變陰性的女性患兒中檢測出位于X染色體上的編碼鈣粘蛋白的PCDH19基因突變[15]。因此,對于女性患兒如SCN1A基因突變檢測陰性,應進行PCDH19基因突變檢測。其他如SCN1B、GABRA1和STXBP1等基因突變導致的DS,亦有報道[16-17]。
對所有抗癲癇藥物抵抗是DS的一個重要特征,目前對DS的治療目標是防止長時間的驚厥發作,減少驚厥發作次數,改善患兒的精神運動發育。因大多數DS患兒攜帶編碼鈉離子通道α亞單位的SCN1A基因突變,鈉離子通道阻滯劑如卡馬西平、奧卡西平、拉莫三嗪、苯妥因可使病情加重,苯妥因還可誘發手足徐動癥,應避免使用。氨己烯酸也可使病情惡化,苯巴比妥和盧非酰胺作用甚微。在疑似DS的首次無熱或復雜性熱性驚厥發作后給予丙戊酸治療目前已經達成共識。DS的病理生理機制被普遍接受的是中間神經元假說,即SCN1A基因突變使抑制性的GABA能神經元被抑制而導致興奮性神經元的過度興奮[18],司替戊醇具有增強GABA能神經元的作用,可以縮短發作時間并預防SE的發生[19],很多文獻報道托吡酯對部分患兒可以減少發作,因此反復驚厥發作可加用托吡酯或司替戊醇,如丙戊酸加托吡酯或司替戊醇效果不佳,可以將托吡酯和司替戊醇互相替換,或替換成溴化物。因為司替戊醇是細胞色素P450的抑制劑,加用司替戊醇時要適當減少丙戊酸的用量。在藥物治療無效的情況下可以選用生酮飲食治療[20]。本報道中2例患兒應用過多種抗癲癇藥物均不能控制發作,在確診DS前例1患兒在托吡酯、左乙拉西坦、丙戊酸鈉治療無效的情況下,曾短時間應用過鈉離子通道阻滯劑奧卡西平。文獻報道約1/3的DS患兒曾有鈉離子通道阻滯劑的應用經歷[21],早期進行SCN1A基因突變檢測不僅可以幫助DS患兒早期明確診斷,還可以避免鈉離子通道阻滯劑等可能加重病情的藥物的應用。2例患兒目前均口服丙戊酸聯合托吡酯治療,癲癇雖未完全控制,但發作次數較前減少,持續時間也較前縮短,均未出現癲癇持續狀態。
通過回顧性分析上述2例患兒的臨床和基因突變特點,可以發現DS患兒在病程中的癲癇頻繁發作期后可以出現強直發作形式;除此以外,2例患兒在病史、臨床表現、對抗癲癇藥物的反應以及預后方面與其他無強直發作的DS患兒基本相同。基因檢測有助于這種少見表型DS的早期確診及選用合適的抗癲癇藥物治療。
[1] Wu YW, Sullivan J, McDaniel SS,etal. Incidence of Dravet syndrome in a US population[J]. Pediatrics, 2015, 136(5): 1310-1315.
[2] Bayat A, Hjalgrim H, Moller RS,etal. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22000: a population-based study from 2004 to 2009[J]. Epilepsia, 2015, 56(4): 36-39.
[3] Shmuely S, Sisodiya SM, Gunning WB,etal. Mortality in Dravet syndrome: a review[J]. Epilepsy Behav, 2016, 64(Pt A): 69-74.
[4] 黃逸青,吳 原.癲癇猝死模型的研究進展[J].醫學研究生學報,2016,29(1):100-103.
[5] Dravet C. Dravet syndrome history[J]. Dev Med Child Neurol, 2011, 53( Suppl 2):1-6.
[6] Scheffer IE. Diagnosis and long-term course of Dravet syndrome[J]. Eur J Paediatr Neurol, 2012, 16( Suppl 1): S5- S8
[7] Dravet C. Les e’pilepsies graves de l’enfant[J]. Vie Med,1978, 8:543-548.
[8] Dravet C. The core Dravet syndrome phenotype[J]. Epilepsia, 2011, 52(Suppl 2):3-9.
[9] Nabbout R, Desguerre I, Sabbagh S,etal. An unexpected EEG course in Dravet syndrome[J]. Epilepsy Res, 2008, 81(1):90-95.
[10] Connolly MB. Dravet syndrome: Diagnosis and long-term course[J]. Can J Neurol Sci, 2016, 43 (Suppl 3): S3-S8.
[11] Gataullina S, Dulac O. From genotype to phenotype in Dravet disease[J]. Seizure, 2017,44:58-64.
[12] Claes L, Del-favero J, ceulemans B,etal. De nove mutation in the sodium channel gene SCN1A cause severe myoclonic epileptic in fancy[J]. Am J Hum genet, 2001, 68(6):1327-1332.
[13] 許小菁, 張月華, 孫慧慧, 等. Dravet綜合征SCN1A基因突變的遺傳特點及表型研究[J].中華醫學遺傳學雜志, 2012, 29(6): 625-630.
[14] Depienne C, Trouillard O, Saint-Martin C,etal. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients[J]. Med Genet, 2009, 46(3): 183-191.
[15] Depienne C, Bouteiller D, Keren B,etal. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females[J]. PLoS Genet, 2009, 5(2):e1000381.
[16] Patino GA, Claes LR, Lopez-Santiago LF,etal. functional null mutation of SCN1B in a patient with Dravet syndrome[J]. J Neurosci, 2009, 29(34):10764-10778.
[17] Carvill GL, Weckhuysen S, McMahon JM,etal. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome[J]. Neurology, 2014, 82(14):1245-1253.
[18] Cheah CS, Yu FH, Westenbroek RE,etal. Specific deletion of NaV1.1 sodium channels ininhibitory interneurons causes seizures and premature death in amouse model of Dravet syndrome[J]. Proc Natl Acad Sci USA, 2012, 109(36):14646-14651.
[19] Grosenbaugh DK, Mott DD. Stiripentol is anticonvulsant by potentiating GABAergic transmission in a model of benzodiazepine-refractory status epilepticus[J]. Neuropharmacology, 2013, 67:136-143.
[20] Wirrell EC. Treatment of Dravet syndrome[J]. Can J Neurol Sci,2016, 43 (Suppl 3): S13-S18.
[21] Aras LM, Isla J, Mingorance-Le Meur A. The European patient with Dravet syndrome: results from a parent-reported survey on antiepileptic drug use in the European population with Dravet syndrome[J].Epilepsy Behav, 2015, 44:104-109.
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
