具有氧空位BixWO6(1.81≤x≤2.01)的第一性原理計算和光催化性能研究?
2018-03-26 19:06:52何金云彭代江王燕舞龍飛鄒正光
物理學報 2018年6期
關鍵詞:產品
何金云 彭代江 王燕舞 龍飛 鄒正光
1)(桂林理工大學,有色金屬及材料加工新技術教育部重點實驗室,桂林 541004)
2)(桂林理工大學,廣西建筑新能源與節能重點實驗室,桂林 541004)
3)(桂林理工大學環境科學與工程學院,桂林 541004)
1 引 言
自1972年Fujishima和Honda[1]發現可以通過在TiO2電極上分解水產生氫氣以來,光催化過程形成的人工光合作用被認為是解決當前的環境和能源問題的希望.實驗證明在氧化物半導體和非氧化物半導體中TiO2是最合適的光催化材料[2],同時具有低成本、環境友好、化學穩定性好且易于制備.但是TiO2有較寬的帶隙,銳鈦礦相3.2 eV,金紅石相3.0 eV,它只能吸收太陽光譜中不到5%的紫外光,而約50%的可見光無法吸收,因此實際應用范圍受限.
主要有兩種方法開發新的光催化劑,一種是對TiO2采用助催化劑進行表面修飾或者通過摻雜來擴展它的吸收邊到可見光區,以提高它對太陽能的利用率[3?6];另一種方法是尋找帶寬合適、吸收范圍在可見光區的新型光催化材料[7].近年來人們發現Bi基氧化物具有可見光吸收帶,價帶頂主要是由Bi 6s和O 2p雜化形成的填滿的反鍵態,從價帶頂到導帶的躍遷所需能量部分位于可見光區[8?11].Bi基氧化物具有無毒、化學和熱力學穩定[12].在Bi基氧化物中,Bi2WO6的禁帶寬度為2.7 eV,能被紫外光和可見光激發.同時,Bi2WO6合成過程中形貌可控、氧化能力強、耐光腐蝕[13?16],作為新型可見光光催化劑應用前景廣闊.
提高Bi2WO6的光催化活性,抑制光生載流子的復合是很重要的.氧空位可作為電子俘獲中心,因而它在抑制光生載流子復合方面具有重要的作用[17?19].而且氧空位還可作為反應物分子的反應中心[20].Zhang等[21]合成了具有氧空位的鋯離子摻雜的Bi2WO6,其光催化性能提高了3倍.Nie等[22]合成了具有氧空位的納米凹片狀Bi2WO6,它具有很高的電催化產氧性能.因此,將氧空位引入到光催化材料中,有望獲得高活性的光催化材料.
本文采用溶劑熱法合成了具有氧空位的BixWO6(1.81≤x≤2.01).采用密度泛函理論(DFT)計算和實驗系統研究了Bi含量和氧空位對Bi2WO6的電子結構、晶體結構和光催化性能的影響.

圖1 包含72個原子的Bi2WO6晶胞和三種氧缺陷的位置Fig.1.Bi2WO6cell with 72 atoms and three kinds of oxygen vacancies.
2 實 驗
2.1 DFT理論計算方法和模型
采用第一性原理計算了氧空位對Bi2WO6晶體結構和電子結構的影響. 采用創騰公司Material Studio軟件中的Castep模塊.計算模型采用包含72個原子的2×1×1 Bi2WO6超胞,原胞晶格參數采用McDowell的實驗值:a=5.488 ?,b=5.4607 ?,c=16.4842 ?, 空 間組群為PCA21[23],模型結構如圖1所示.采用Broyden-Fletcher-Goldfarb-Shanno優化算法;交換-關聯勢采用廣義梯度近似中的PBESOL泛函[24]來處理電子與電子之間的相互作用交換相關能,對Bi,O,W加超軟贗勢來描述價電子與離子實之間的相互作用,計算時分別將Bi的6s26p3,O的2s22p4,W的5d46s2軌道電子作為價電子,電子波函數用平面波基組展開,平面波截斷能經過收斂測試后設為380 eV,體系的總能量和電荷密度計算在K空間布里淵區積分通過Monkhorst-Pack方法產生的3×6×2點網絡下進行,保證了體系能量和構型在準完備平面波基組水平上的收斂.對幾何結構進行優化的收斂標準為:體系總能量變化≤5×10?6eV/atom,原子受力≤0.05 eV/?,原子間內應力≤0.1 GPa.在自洽場運算中,電子最小化采用密度混合法,密度混合采用了Pulay混合法,自洽場收斂精度為5×10?6eV/atom.能帶計算時在K空間沿以下高對稱點進行:G(0,0,0),F(0,0.5,0),Q(0,0.5,0.5)和Z(0,0,0.5).原子和成鍵布居分析采用Mulliken布居分析.如圖1所示,在Bi2WO6超胞中分別有三種O缺陷位,分別標記為O1,O2,O3,計算過程中固定晶胞晶格參數,只優化原子位置.
2.2 具有氧空位BixWO6(1.81≤x≤2.01)光催化劑的合成
采用溶劑熱法合成具有氧空位的非化學計量BixWO6(x=1.81,1.87,1.89,1.92,2.01)光催化劑.具體過程如下:將10 mmol Na2WO4·5 H2O溶解于30 mL去離子水中.同時,將不同質量的Bi(NO3)3·3H2O(18.5,19,19.5,20和20.5 mmol)80°C溶解于30 mL乙酸中,將這兩種溶液混合后攪拌1 h.然后將這五組混合液倒入100 mL水熱釜中,放入烘箱,于180°C反應15 h.自然冷卻到室溫后,分別將所得到的沉淀物用去離子水和無水乙醇離心洗滌3次,于80°C烘干,得到BixWO6(x=1.81,1.87,1.89,1.92,2.01)產品,并標記為BWO-X(X=1,2,3,4,5).
3 結果與討論
3.1 DFT計算
3.1.1 Bi2WO6?y的氧空位缺陷形成能
缺陷形成能是衡量一個體系是否容易形成以及結構相對穩定性的重要指標,根據下式計算缺陷形成能Ef:
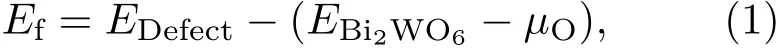
其中,Ef為形成能,Edefect為缺陷體系的能量,EBi2WO6為未摻雜Bi2WO6體系的能量,μO為O原子的化學勢,計算結果如表1所列.

表1 具有一個氧空位缺陷的Bi16W8O48的形成能Table 1.Formation energy of Bi16W8O48with one oxygen vacancy.
形成能越小,說明缺陷越容易形成,反之則越不容易形成.由表1可知O1位缺陷最有可能形成,而O3缺陷的形成難度最大.
3.1.2 氧空位缺陷態Bi2WO6?y的原子間鍵長和布居分析
原子間鍵長和電荷布居的計算結果表明,氧空位缺陷主要影響與之有化學鍵作用的外兩層電子,因此主要選擇分析與Bi原子相鄰的6個O原子位置及與其成鍵的Bi,W原子,氧空位缺陷形成前后相應的原子間鍵長如圖2.
從圖2可以看出:形成O1位缺陷后,主要影響[Bi2O2]2+層,但總體影響不大,晶體內部其他原子間鍵長變化也不明顯,晶格畸變較小;形成O2位缺陷后,主要影響[Bi2O2]2+和[WO6]6?層間,但總體影響不大,只引起缺陷周圍W—O,Bi—O之間鍵長輕微減小,晶體內部其他原子間鍵長變化不明顯,晶格畸變較小;形成O3位缺陷后,主要影響[WO6]6?層內的W,O原子,只引起缺陷周圍W—O鍵長輕微減小,晶體內部其他原子間鍵長變化不明顯,晶格畸變較小.因此,三種位置的氧缺陷引起的晶格畸變都較小,對Bi2WO6的晶體結構無明顯影響.
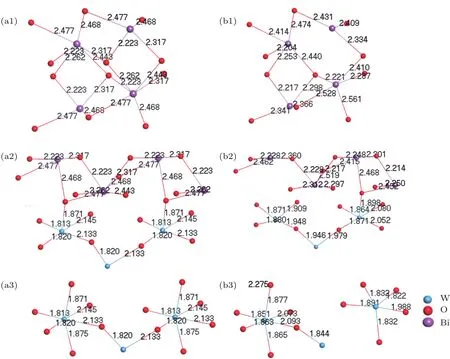
圖2 無缺陷及有一個氧空位缺陷Bi16W8O48晶胞的鍵長(單位?) (a1)無缺陷O1位;(b1)缺陷O1位;(a2)無缺陷O2位;(b2)缺陷O2位;(a3)無缺陷O3位;(b3)缺陷O3位Fig.2.Bond length of Bi16W8O48cell with or without one oxygen vacancy(unit:?):(a1)Without O1 oxygen vacancy;(b1)with O1 oxygen vacancy;(a2)without O2 oxygen vacancy;(b2)with O2 oxygen vacancy;(a3)without O3 oxygen vacancy;(b3)with O3 oxygen vacancy.
圖3和表2直觀地顯示了形成氧空位缺陷后原子電荷布居的變化.可以看出,O1空位形成后,缺陷位相鄰Bi原子電荷布居明顯減小,氧原子電荷基本保持不變,Bi—O鍵布居減小,共價性減弱,主要是由于氧原子電負性較強,形成空位缺陷后周圍Bi原子所需提供電子數相應減小;其他Bi—O鍵影響不大,共價性略微增強.形成O2空位后,缺陷周圍Bi,W原子電荷布居均減小,但W原子周圍的氧原子電荷數增加,總體上鍵長減小、鍵布居增加,原子間共價性作用增強.形成O3位缺陷后,W原子電荷布居減小,主要是由于減少了一個W—O鍵,缺陷位相鄰W原子周圍的氧原子電荷數增加,W—O鍵布居增加,共價性增強,總體上導致W—O間作用增強.計算結果說明,氧空位使Bi2WO6的晶體結構生成了更多的缺陷,一定數目的晶體缺陷可有效俘獲光生電子,抑制電子-空穴對的復合,從而提高半導體的光催化活性.
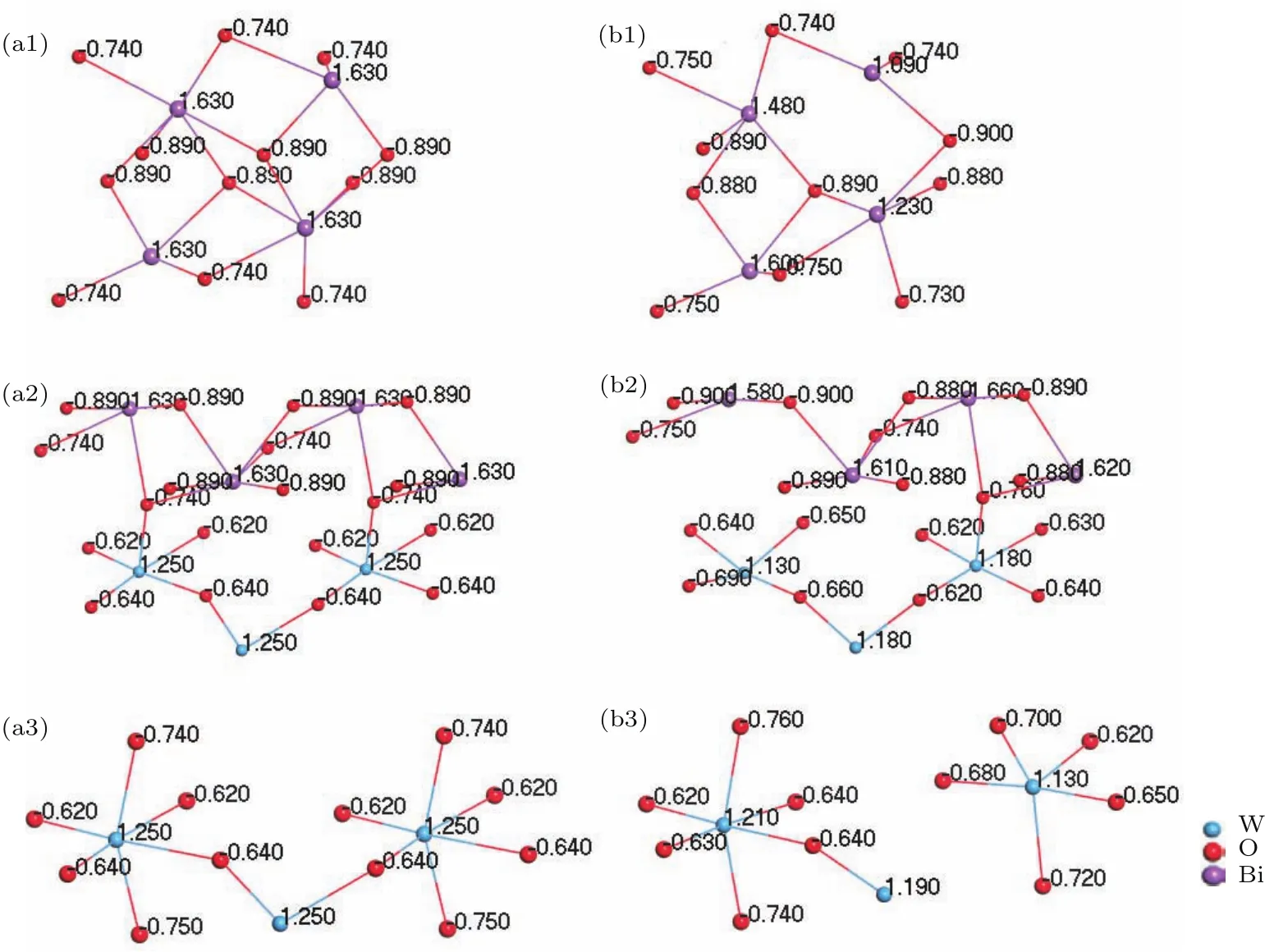
圖3 無缺陷及有一個氧空位缺陷的Bi16W8O48晶胞電荷布居 (a1)無缺陷O1位;(b1)缺陷O1位;(a2)無缺陷O2位;(b2)缺陷O2位;(a3)無缺陷O3位;(b3)缺陷O3位Fig.3.Charge population of Bi16W8O48cell with or without one oxygen vacancy:(a1)Without O1 oxygen vacancy;(b1)with O1 oxygen vacancy;(a2)without O2 oxygen vacancy;(b2)with O2 oxygen vacancy;(a3)without O3 oxygen vacancy;(b3)with O3 oxygen vacancy.
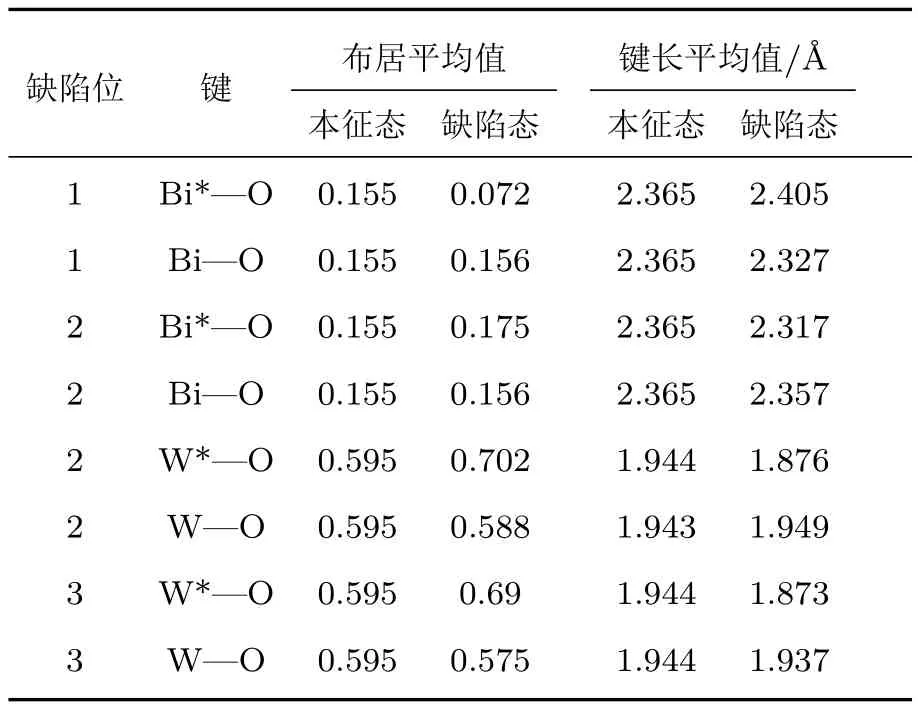
表2 氧空位附近的原子鍵布居和鍵長Table 2.Bond population and bond length of atom near oxygen vacancy.
3.1.3 氧空位缺陷態Bi2WO6?y的電子結構分析
由圖4形成氧空位缺陷后的能帶結構及圖5總態密度圖可以看出,三種氧空位均導致體系禁帶寬度變窄,導帶底和價帶頂能量均降低,與文獻[25]的實驗結果一致.其中O1位帶隙降低是由于禁帶中間形成了雜質能級,主要是Bi的6s軌道和O的2p軌道雜化引起,可能是因為Bi—O間電子相互作用增強所致.O2,O3位帶隙降低是由于W—O間電子相互作用增強導致導帶底能級離散,同時W的5d和O的2p雜化軌道能量降低引起.由于形成空位后費米能級均位于導帶底,因此都是n型半導體.由此可見,三種氧空位都會導致體系帶隙變窄,使光生電子躍遷更加容易,有利于提高Bi2WO6的光催化性能.由此,我們采用溶劑熱法合成了具有氧空位的BixWO6(1.81≤x≤2.01)產品,以獲得高活性可見光催化材料.
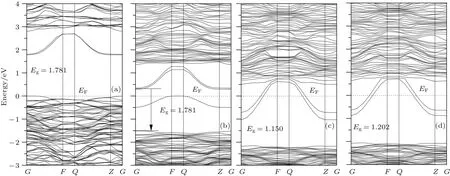
圖4 本征態及形成氧空位缺陷后的Bi16W8O48晶胞能帶結構圖 (a)本征態;(b)O1位空位;(c)O2位空位;(d)O3位空位Fig.4.Calculated band structure of Bi16W8O48cell:(a)Eigenstate;(b)oxygen vacancy at O1;(c)oxygen vacancy at O2;(d)oxygen vacancy at O3.
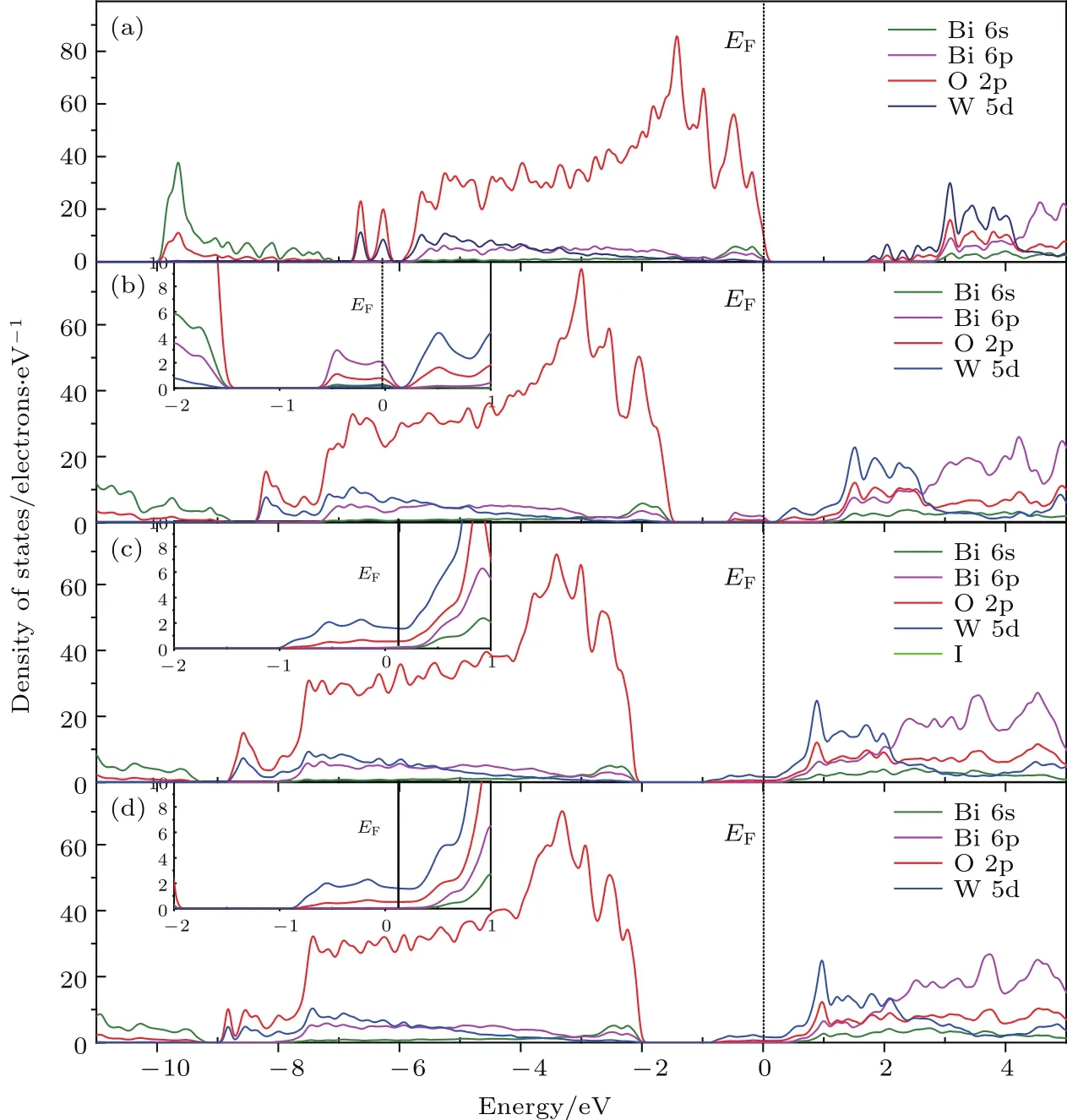
圖5 本征態及形成O空位缺陷后的Bi16W8O48晶胞總態密度圖 (a)本征態;(b)O1位空位;(c)O2位空位;(d)O3位空位Fig.5.Total density of states of Bi16W8O48cell:(a)Eigenstate;(b)oxygen vacancy at O1;(c)oxygen vacancy at O2;(d)oxygen vacancy at O3.
3.1.4 氧空位缺陷態Bi2WO6?y的吸收光譜分析
從晶胞吸收光譜圖(圖6)可看出,形成氧空位缺陷后,晶體在可見光區吸收強度都有所增加,其中O1空位情況下增加較多.原因主要是形成氧空位缺陷后,體系的帶隙減小.O1空位可見光區吸收強度增加較多的原因是O1空位還形成了雜質能級.結果表明,具有氧空位的半導體的光催化活性較沒有氧空位的高.
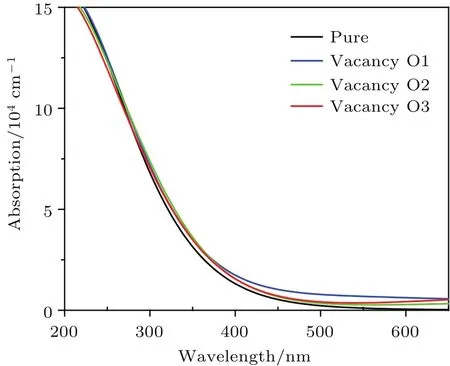
圖6 本征態及形成氧空位缺陷后Bi16W8O48晶胞的吸收光譜圖Fig.6.Absorption spectra of eigen Bi16W8O48cell and Bi16W8O48cell with oxygen vacancy defects.
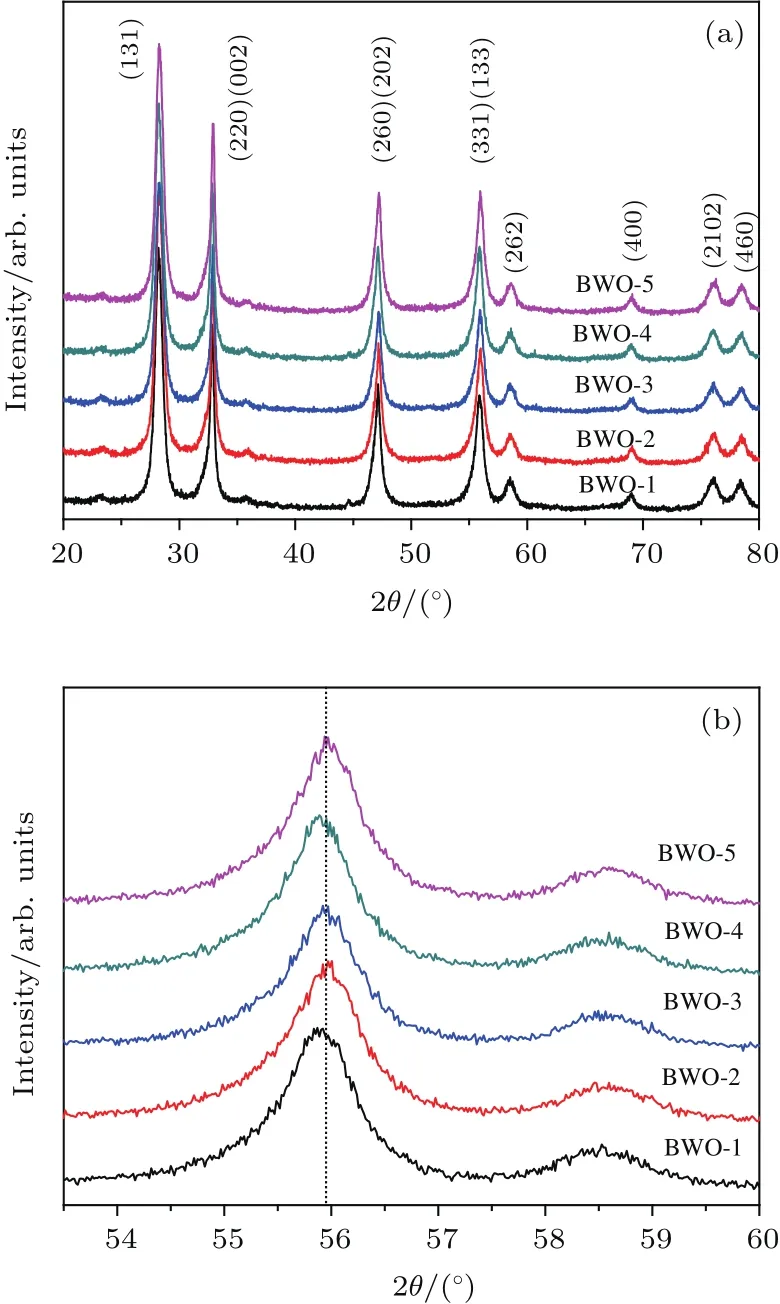
圖7 BixWO6產品的X射線衍射譜圖 (a)全譜圖;(b)放大圖Fig.7. XRD patterns of the BixWO6products:(a)Full spectra,(b)enlarged patterns.
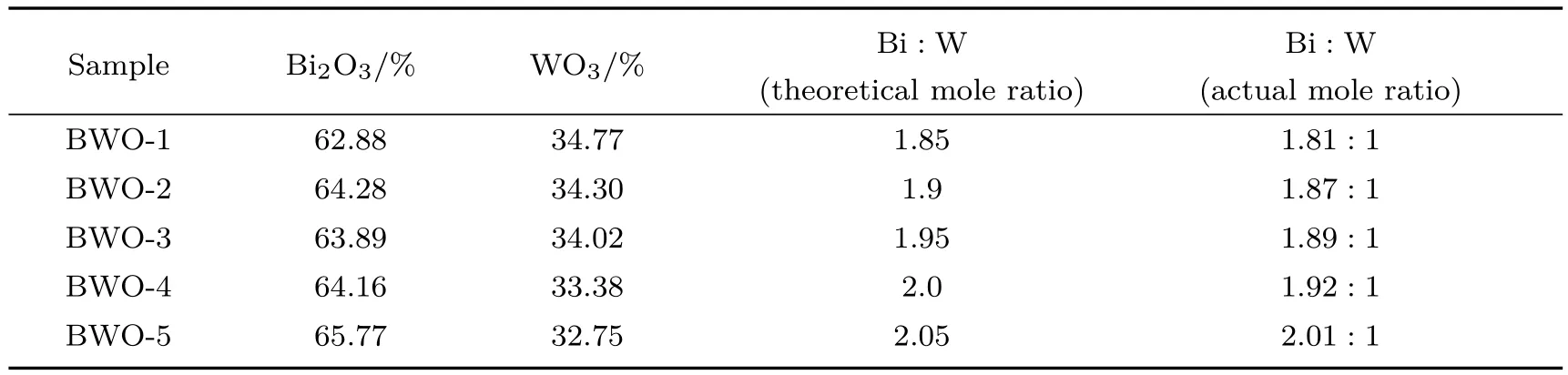
表3 BixWO6產品的XRF結果Table 3.XRF results of the BixWO6products.
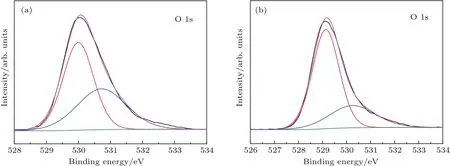
圖8 (a)BWO-3和(b)BWO-5產品的XPS高分辨O 1s圖譜Fig.8.High-resolution XPS spectra of O 1s for(a)BWO-3 and(b)BWO-5 products.
3.2 BixWO6(1.81≤x≤2.01)的實驗研究
3.2.1 BixWO6(1.81≤x≤2.01)產品的相組成
圖7為BWO-X(X=1,2,3,4,5)產品的X射線衍射(XRD)譜.從圖7可以看出,所有產品的XRD衍射峰都屬于正交相Bi2WO6(JCPDS No.39-0256),沒有發現其他物相的衍射峰,說明所有產品都是純的Bi2WO6產品.表3為產品的X射線熒光光譜(XRF)分析數據,可以看出,所有產品的Bi,W摩爾比都較它們的理論值低.BWO-5產品的Bi,W摩爾為2.01,與Bi2WO6的理論化學計量比相近.
采用X射線光電子能譜(XPS)研究了BWO-3和BWO-5產品氧空位的存在情況(圖8). 從BWO-3和BWO-5產品的O 1s譜可以看出,它們都由一個低結合能峰和一個高結合能峰組成,這兩個結合能峰分別歸屬于晶格氧和化學吸附氧,如羥基和H2O[26],其中高結合能O 1s峰的面積隨著氧空位的增加而增加[27].BWO-3和BWO-5產品的高結合能O 1s峰的面積比為1.18,說明BWO-3中存在較多的氧空位.由DFT計算結果可知,氧空位會導致Bi2WO6的帶隙變窄,使光電子躍遷更加容易,因而BWO-3產品具有更好的光催化性能.
3.2.2 合成BixWO6(1.81≤x≤2.01)產品的顯微結構
產品的形貌和微觀結構通過場發射掃描電子顯微鏡(FESEM)和透射電子顯微鏡(TEM)分析得到.從BWO-X(X=1,2,3,4,5)產品的FESEM圖可以看出,所有產品的形貌都由花狀微球和納米片組成,花狀微球的平均粒徑為2—3μm(圖9).從BWO-3產品的放大圖可以看出,這些微球由一些納米片構成.從BWO-3樣品的TEM圖可知,這些納米片由尺寸小于100 nm的納米薄片組裝而成(圖10(a)).這些納米薄片上的HRTEM圖可看到清晰的寬度為0.27 nm的晶格條紋(圖10(b)),歸屬于Bi2WO6的(006)晶面.
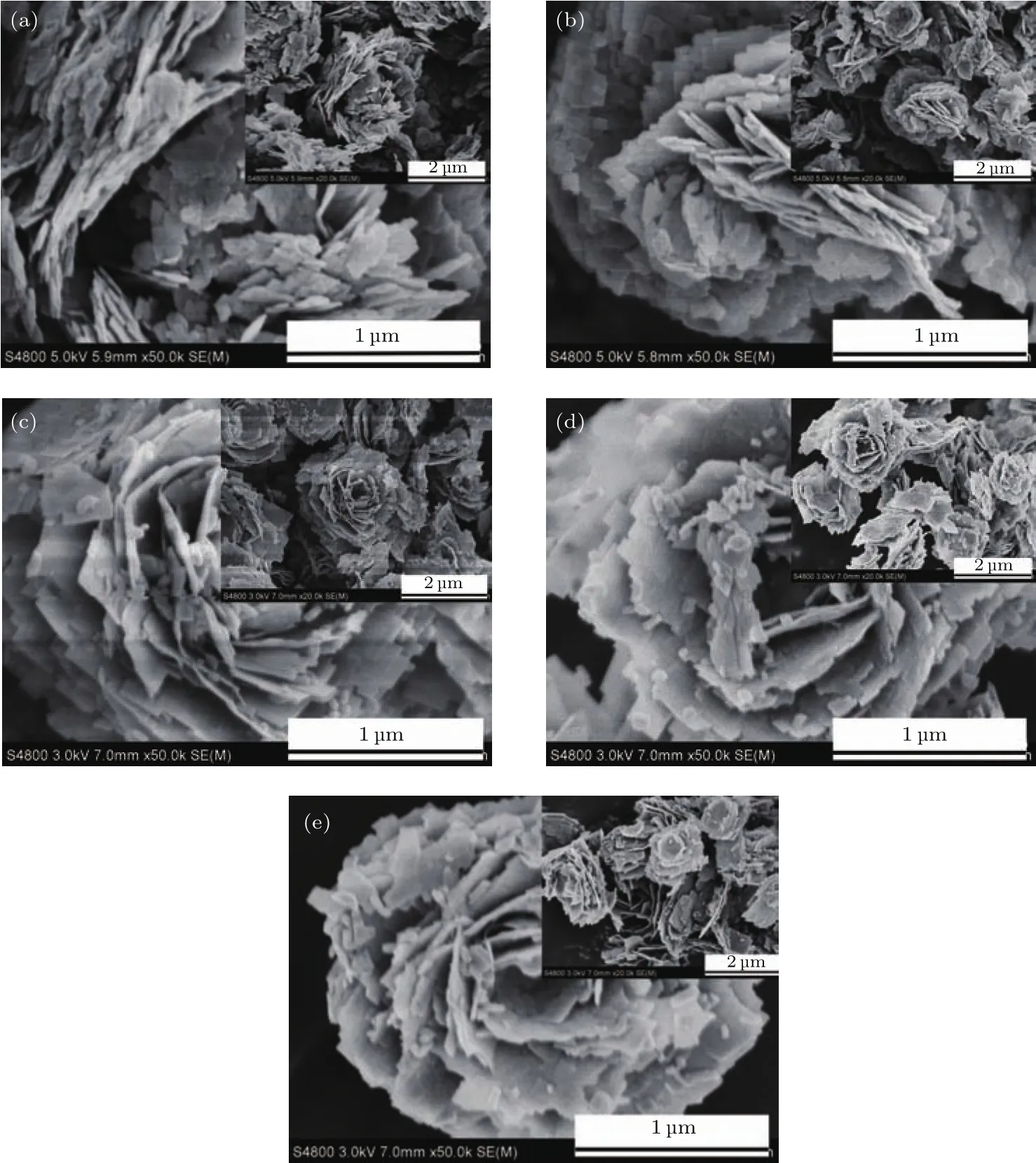
圖9 產品的FE-SEM圖像 (a)BWO-1;(b)BWO-2;(c)BWO-3;(d)BWO-4;(e)BWO-5Fig.9.FE-SEM images of the products:(a)BWO-1;(b)BWO-2;(c)BWO-3;(d)BWO-4;(e)BWO-5.

圖10BWO-3產品的(a)TEM和(b)HRTEM圖像Fig.10.(a)TEM and(b)HRTEM image of the BWO-3 product.
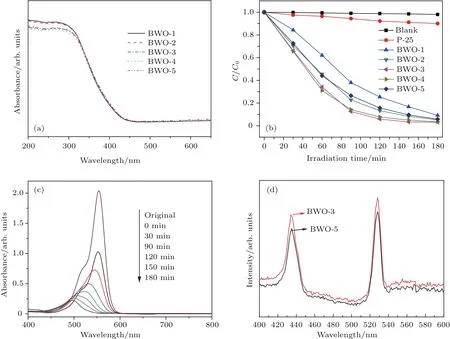
圖11 (a)樣品的紫外-可見吸收光譜;(b)樣品可見光催化降解RhB溶液(1×10–5mol/L)的性能;(c)以BWO-3產品為催化劑,可見光(λ>420 nm)照射下,RhB水溶液隨光照時間變化的吸收光譜;(d)BWO-3和BWO-5產品的熒光光譜Fig.11.(a)UV-visible diffuse re fl ectance spectra of the samples;(b)UV-visible spectral changes of RhB(1×10?5 mol/L)in aqueous catalyst dispersions as a function of irradiation time;(c)the temporal evolution of the spectrum during the photodegradation of RhB mediated by BWO-3 catalyst under visible light illumination(λ> 420 nm);(d)PL spectra of BWO-3 and BWO-5 product.
3.2.3 紫外可見光吸收譜和光催化性能
影響半導體光催化性能的另一關鍵因素是其能帶結構[28].圖11(a)為產品的紫外-可見光吸收譜.從圖11(a)可以看出,所有樣品的光吸收邊都從紫外區延伸到460 nm處的可見光區,說明這些樣品都具有可見光催化活性,實驗結果與圖6的DFT計算結果相一致.在這些產品中,BWO-3產品在可見光區的吸收最強,預示了其最好的光催化性能.
為了表征BWO-X(X=1,2,3,4,5)產品的光催化性能,研究了可見光(λ>420 nm)照射下樣品催化降解羅丹明B(RhB)水溶液的性能.圖11(b)為以BWO-X(X=1,2,3,4,5)為催化劑時,不同光照時間RhB的降解率.C0和C分別為光照前和光照一定時間后RhB的濃度.在同等降解實驗條件下,以未放催化劑的空白實驗和TiO2(P-25)催化劑作為對比實驗.從圖11(b)可以看出,光照180 min后,未放催化劑的空白實驗中,RhB幾乎未發生降解,說明RhB的光穩定性很好.而使用P-25為催化劑時,RhB只降解了8%.而本文的樣品中,BWO-3樣品的光催化性能最好,RhB降解了98%.BWO-4樣品的光催化性能略差于BWO-3樣品.而BWO-2和BWO-5樣品由于其Bi,W比相差不大,因此它們的光催化性能也相差不大,RhB都降解了96%.當使用BWO-3為催化劑,光照時間不同時,RhB溶液的光吸收譜的變化如圖11(c)所示.RhB主要的特征吸收峰位于553 nm波長處,光照射180 min后,RhB幾乎被完全降解,其乙基基團被氧化脫除,染料的顏色也由玫瑰紅變為無色.
由于熒光發射主要來源于半導體中自由載流子的復合,因而熒光發射光譜常被用來表征載流子復合率的高低.材料的光生電子和空穴的復合速率越高,其熒光激發光譜強度越高.光生電子和空穴的復合率是影響光催化活性的主要因素,復合率越小,光催化活性越高.圖11(d)為BWO-3和BWO-5的熒光光譜圖,BWO-3的熒光強度較BWO-5的弱,這與其光催化性能較好是相一致的.這是因為,非化學計量導致BWO-3產品中存在較多的氧空位,生成帶正電荷的VO缺陷,它們可作為俘獲中心俘獲光生電子,減少光生電子和空穴對的復合[29].
從以上DFT計算、實驗研究的結果分析可知,BWO-3產品光催化性能最好的原因如下:一方面,非化學計量導致BWO-3產品中存在較多的氧空位,生成帶正電荷的VO缺陷,它們可作為俘獲中心俘獲光生電子,減少光生電子和空穴對的復合[29];元素的非化學計量也可以促進光生電子的產生,俘獲水分子,生成更多的O2?[30].由DFT計算可知,產生氧空位后,氧空位缺陷導致Bi2WO6的帶隙變窄,使光電子躍遷更加容易;同時,氧空位使Bi2WO6的晶體結構生成更多的缺陷,一定數目的晶體缺陷可有效俘獲光生電子,抑制電子-空穴對的復合.因此,Bi元素的非化學計量和氧空位的協同作用使BWO-3產品的光催化性能最好.
4 結 論
采用溶劑熱法合成了具有氧空位的非化學計量BixWO6(x=1.81,1.87,1.89,1.92,2.01)光催化劑.DFT計算結果表明,氧空位的存在可顯著減小Bi2WO6的帶隙,有利于光生電子的生成,并有效抑制電子空穴的復合.實驗結果表明,Bi元素的含量小于化學計量比時,Bi2WO6的晶體結構發生了微小變形,Bi元素含量對Bi2WO6的顯微結構影響不大,但會影響產品氧空位的含量和光吸收性能.Bi1.89WO6產品的光催化性能最佳,可見光照射180 min后,可降解98%的RhB.采用適當方法,使產品具有氧空位和非化學計量是獲得高光催化活性材料的一種有效方法.
[1]Fujishima A,Honda K 1972Nature238 37
[2]Jing L,Sun X 2003Sol.Energy Mater.Sol.Cells79 133
[3]Liu Y,Yu L,Wei Z G,Pan Z C,Zou Y D,Xie Y H 2013Chem.J.Chin.Univ.(in Chinese)[劉月,余林,魏志鋼,潘湛昌,鄒燕娣,謝英豪2013高等學校化學學報34 434]
[4]Carp O,Huisman C L,Reller A 2004Prog.Solid State Chem.32 33
[5]Yang K,Dai Y,Huang B 2008Chem.Phys.Lett.456 71
[6]Wang P,Huang B,Lou Z 2010Chem.Eur.J.16 538
[7]Kubacka A,Fern Ndezgarc A M,Col N G 2012Chem.Rev.112 1555
[8]Kudo A,Omori K,Kato H 1999J.Am.Chem.Soc.121 11459
[9]Fu H,Pan C,Yao W 2005J.Phys.Chem.B109 22432
[10]Zhang L,Wang W,Yang 2006J.Appl.Catal.A308 105
[11]Lai K,Zhu Y,Lu J 2013Comput.Mater.Sci.67 88
[12]Zeng D W,Xie C S,Zhu B L 2003Mater.Sci.Eng.B104 68
[13]Zhang L,Wang W,Zhou L 2007Small3 1618
[14]Zhang Z,Wang W,Gao E 2012J.Phys.Chem.C116 25898
[15]Bhattacharya C,Lee H C,Bard A J 2013J.Phys.Chem.C117 9633
[16]Sun Z X,Li X F,Guo S,Wang H Q,Wu Z B 2013J.Colloid Interf.Sci.412 31
[17]Kuo T J,Lin C N,Kuo C L,Huang M H 2007Chem.Mater.19 5143
[18]Wang J C,Liu P,Fu X Z,Li Z H,Han W,Wang X X 2009Langmuir.25 1218
[19]Zheng Y H,Chen C Q,Zhan Y Y,Lin X Y,Zheng Q,Wei K M,Zhu J F,Zhu Y J 2007Inorg.Chem.46 6675
[20]Gong X Q,Selloni A,Batzil M 2006Nat.Mater.5 665
[21]Zhang Z,Wang W,Gao E,Shang M,Xu J 2011J.Hazard Mater.196 255
[22]Nie Z,Ma D,Fang G Y,Chen W,Huang S M 2016J.Mater.Chem.A4 2438
[23]Mcdowell N A,Knight K S 2006Chem.Eur.J.12 1493
[24]Perdew J P,Ruzsinszky A,Csonka G I 2008Phys.Rev.Lett.101 136406
[25]Lu Q,Hua L G,Chen Y L 2015J.Inorg.Mater.30 413(in Chinese)[盧青,華羅光,陳亦琳2015無機材料學報30 413]
[26]Zhou B,Zhao X,Liu H 2010Appl.Catal.B99 214
[27]Sun S B,Chang X T,Li Z J 2012Mater.Charact.73 130
[28]Lin Z,Wang W,Liu S 2006J.Mol.Catal.A252 120
[29]Wu J,Duan F,Zheng Y 2007J.Phys.Chem.C111 12866
[30]Ding X,Zhao K,Zhang L 2014Environ.Sci.Technol.48 5823
猜你喜歡
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
物流技術與應用(2022年5期)2022-06-17 06:01:38
快樂語文(2021年36期)2022-01-18 05:48:46
金橋(2021年4期)2021-05-21 08:19:22
中國化妝品(2018年6期)2018-07-09 03:12:40
中國化妝品(2018年6期)2018-07-09 03:12:32
Coco薇(2015年1期)2015-08-13 02:23:50
汽車維修與保養(2015年6期)2015-04-17 03:31:50
玩具(2009年10期)2009-11-04 02:33:14
