N催化劑中活性組分的核磁共振氫譜定量研究
2018-03-06 08:22:50黃文氫
分析測試學報 2018年2期
關鍵詞:催化劑
殷 杰,黃文氫
(中國石油化工股份有限公司 北京化工研究院,北京 100013)
從第一代Ziegler-Natta (Z-N) 催化劑發展到目前的高效載體型催化體系,作為調節劑的給電子體是催化劑研究的核心[1]。給電子體化合物在Z-N催化劑中的作用主要有:使無規活性中心失活,使無規活性中心轉化為等規活性中心,以及提高等規活性中心的鏈增長常數等。按照加入方式的不同,給電子體化合物可分為內給電子體和外給電子體。其中,內給電子體在催化劑制備時加入,外給電子體在丙烯聚合時加入。內外給電子體的獨立性與互補性影響著聚丙烯催化劑的性能。內給電子體對于載體化的Z-N催化劑發揮了最核心的作用[2-3]:一方面與MgCl2絡合,改變了催化劑的催化活性和定向性;另一方面,控制了TiCl4在MgCl2上的數量和分布,防止產生無規活性中心。內給電子體按其結構的不同可分為單酯類、雙酯類、二醇酯類、二醚類、二酮類等。
N催化劑是北京化工研究院自主研發的以MgCl2為載體的第四代高效聚丙烯催化劑,通過前期研究可知[4-5],N催化劑中除了可以加入鄰苯二甲酸正丁酯(DNBP)、鄰苯二甲酸正丁酯(DIBP)等內給電子體外,催化劑本身在制備時原料間也發生反應產生了活性組分,在滴加TiCl4之前體系中產生了PA-DCP-MgCl和DCP-MgCl兩種給電子體,這些給電子體的存在影響著N催化劑的顆粒形態及催化性能。因此建立該類給電子體含量的測定方法,對于催化劑機理研究及性能改進具有重要意義。
核磁共振法廣泛應用于有機化合物的結構解析和定性分析[6],在化合物純度定值、含量測定中具有很多優勢[7-9]。定量核磁共振法以含氫有機化合物的NMR波譜信號積分面積與氫原子數目成正比為依據,不需引進任何校正因子,不需以每一種被測物的純品作為參比標準。另外,本文研究對象包含的其中兩種給電子體PA-DCP-MgCl和DCP-MgCl,均通過化學反應所得,若采用傳統液相色譜的定量方法,無市售標準品可得,給實驗增加困難。本文采用核磁共振氫譜內標法,建立了一種快速、專屬、簡單,用于無對照品的N催化劑中活性組分含量定量的表征方法。
1 實驗部分
1.1 儀器與原料
超導核磁共振譜儀:AVANCE 300型,瑞士布魯克公司。
無水MgCl2、磷酸三丁酯(TBP)、環氧氯丙烷(ECP)、鄰苯二甲酸酐(PA);N催化劑:工業級,中國石化股份有限公司催化劑北京奧達分公司提供;無水MgCl2經研磨后使用,TBP和ECP經4A分子篩干燥后使用;對苯二酚(HQ,純度99.5%),氘代甲醇(Methanol-d4,純度99.8%),氘代甲苯(Toluene-d8,純度99.5%),均購于百靈威科技有限公司。
1.2 制備方法
鄰苯二甲酸酐溶解液的制備:惰性氣體保護下,向反應器中加入0.6 mL氘代甲苯、30 mg無水MgCl2、78.2 μL TBP和 25.5 μL ECP,在56 ℃下反應2.5 h后,加入8.8 mg PA,反應1 h,形成鄰苯二甲酸酐溶解液體系。取0.5 mL該反應液置入5 mm的核磁管中,然后放入核磁譜儀探頭中進行氫譜測試。
N催化劑按文獻[10]的方法制備,添加的內給電子體為DNBP。
1.3 1H NMR表征條件
測定溫度:25 ℃,環境濕度:35%,觀察頻率:300.13 MHz,譜寬:6 172.84 Hz,探頭溫度298 K,時間域數據點64 k,90°脈沖寬度為12.2 μs,采樣時間為5 s,脈沖延遲時間為12 s,累加次數為32。
1.4 實驗方法
分別準確稱取適量N催化劑和內標物置于直徑5 mm核磁共振樣品管中,加適量氘代甲醇振蕩溶解,制成待測試樣溶液。在上述實驗條件下調整儀器參數,調諧探頭、勻場、采樣,得到1H NMR 譜。再進行相位和基線調整,對N催化劑和內標物的定量峰分別進行5次積分,取其平均值,得到積分結果。
以1H NMR內標法,按下式計算體系內活性組分含量[11-12]。
其中:As為被測樣品定量峰的積分面積,ns為被測樣品定量峰包含的質子數,Ms為被測樣品的分子量,Ar為內標物定量峰的積分面積,nr為內標物定量峰包含的質子數,Mr為內標物的分子量,mr為內標物質量,Wr為內標物的質量分數,ms為樣品質量。
2 結果與討論
2.1 N催化劑中活性組分的結構確認及溶劑的選擇
N催化劑的制備主要包括4個步驟:(1)無水MgCl2溶解于ECP、TBP和甲苯體系的過程,(2)PA的溶解過程,(3)滴加TiCl4析出活化的MgCl2的過程,(4)酯和鈦負載處理、洗滌。
為了對N催化劑制備過程中MgCl2溶解過程和PA溶解過程中產生的新化合物進行化學結構確認,按照“1.2”方法制備了鄰苯二甲酸酐溶解液。該溶解液在氘代甲苯中的核磁共振氫譜分辨率不佳(圖1B),影響數據的進一步分析。通過對不同氘代溶劑中氫譜的比對分析,將體系中的氘代甲苯減壓除去后加入氘代甲醇,得到峰形尖銳、分辨率佳的氫譜圖(圖1A)。通過譜峰化學位移和積分面積的分析,4組峰dH、cH、bH、aH歸屬于原料TBP;積分面積比eH∶fH = 4∶1歸屬于ECP開環后產物DCP-MgCl;積分面積比hH∶iH∶jH∶gH∶hH = 1∶1∶2∶1∶4,歸屬于PA酸酐環斷裂后生成的鄰苯二甲酸酯類產物PA-DCP-MgCl。
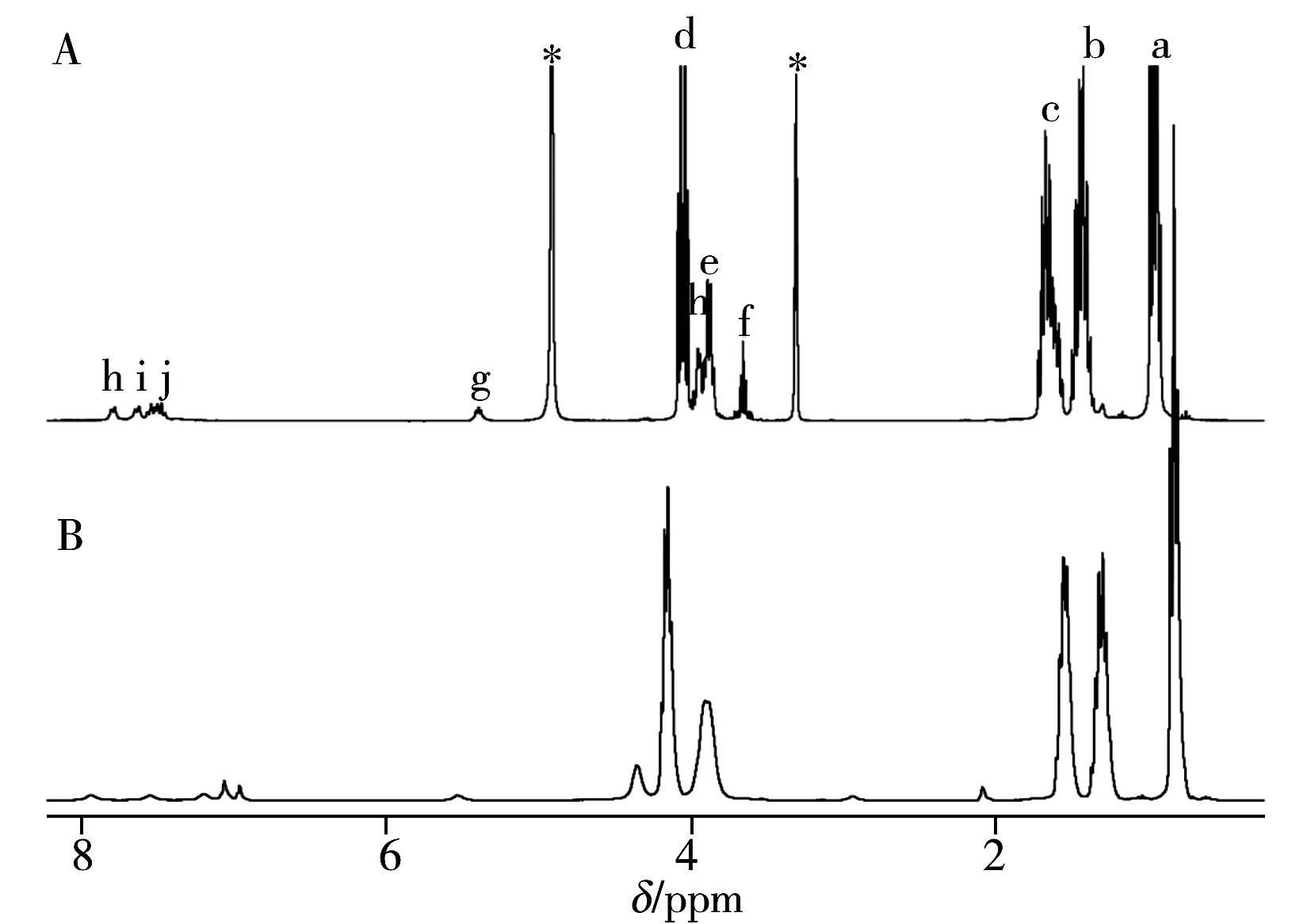
圖1 鄰苯二甲酸酐溶解液在不同溶劑中的1H NMR譜圖Fig.1 1H NMR spectra of PA reaction mixture in different solventsA:methanol-d4;B:toluene-d8

圖2 N催化劑核磁共振氫譜內標法中內標物的選擇Fig.2 1H NMR spectrum of N catalyst with HQ as internal standard
2.2 內標物及定量峰的選擇
內標物質應具有較高的純度和準確含量,不與樣品和溶劑發生化學反應或締合,能溶于氘代溶劑,含有多個質子,具有易于識別的譜峰等特點,并且內標峰與樣品的定量峰能達到基線分離[13]。
根據之前的研究經驗[4-5,14],選擇對苯二酚為內標物,如圖2箭頭所示,對苯二酚在δ= 6.6處有一尖銳的單峰,對應于苯環上的4個氫,并與待測物譜峰均無重疊,可作為內標物的定量峰。如圖3所示,對于N催化劑中待測的4種活性組分TBP、PA-DCP-TiClx、DCP-TiClx和DNBP,分別選取dH、hH、eH、kH作為它們的定量峰,這幾組定量峰峰形尖銳并完全分離,且可與內標物的定量峰明顯區分,滿足核磁氫譜內標法定量的要求。
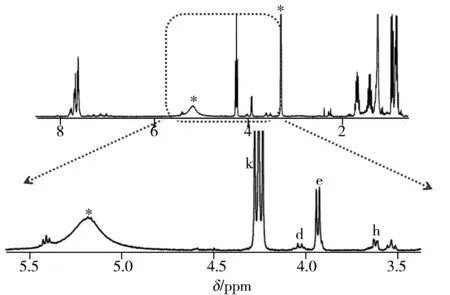
圖3 N催化劑核磁共振氫譜內標法中定量峰的選擇Fig.3 Selection of quantitative peaks in 1H NMR spectrum of N catalyst
2.3 采樣時間的選擇
核磁譜圖是通過采集自由衰減信號(FID)并經傅立葉變換而得到,因此需足夠的采樣時間以保證自由感應衰減能衰減完全得到分辨率較好的譜圖。但過長的采樣時間會導致實驗時間大大增加,實驗中以不同的采集時間采集圖譜,結果表明內標物和樣品的FID信號均在5 s以內衰減完全,因此將采樣時間選定為5 s。
2.4 脈沖延遲時間的選擇
核磁定量研究中,延遲時間是最重要的參數之一。一般來講,延遲時間(D1)為2~5倍于被測物的縱向弛豫時間(T1),延遲時間過短可能造成核的飽和現象,使測定結果偏差較大,而過長的延遲時間會大大增加測試時間[15]。
實驗以kH作定量峰為例,按從短(1 s)到長(25 s)分別測定樣品的1H NMR,考察被測物定量峰與內標物峰面積之比As/Ar隨延遲時間的變化。結果顯示,當延遲時間小于12 s時,As/Ar的值隨延遲時間的增加而增加,而當延遲時間進一步增加時,As/Ar的值基本不變,因此實驗選取D1=12 s。
2.5 定量結果
分別稱取5份N催化劑樣品,按“1.4”方法,在選定的實驗條件下測試1H NMR圖譜,并對選取的定量峰分別進行積分,按1H NMR內標法公式計算樣品純度。經計算,N催化劑中4種活性組分TBP、PA-DCP-TiClx、DCP-TiClx和DNBP在催化劑中的摩爾含量分別為0.008、0.013、0.043、0.209 mmol/g。由于N催化劑在制備過程中滴加TiCl4時,除產生一元取代產物外,還可能存在二元、三元或四元取代產物,該體系較復雜,本文將反應后有機產物中的Ti以—TiCl3基團的存在來計算N催化劑中待測的活性組分含量。
稱取N催化劑試樣和對苯二酚標準樣品適量,在同一實驗條件下連續做6次1H NMR譜,分別選取其中的kH、dH、eH、hH定量峰與內標定量峰積分面積比進行計算(表1),其相對標準偏差(RSD)分別為0.35%、0.75%、0.41%和0.89%,表明該實驗方法的重復性較好。
分別稱取5份同一批次的N催化劑試樣,按“1.3”方法,在選定的實驗條件下做1H NMR圖譜,并分別對選取的定量峰和內標峰進行積分,按1H NMR內標法公式計算其中有效組分的含量。結果表明,其中活性組分TBP、PA-DCP-TiCl3、DCP-TiCl3和DNBP的質量分數分別為0.21%、0.58%、1.21%和5.82%,占N催化劑總量的7.82%。在實驗中,采用高效液相色譜(HPLC)法以甲醇-水為流動相對該批次的N催化劑樣品中的DNBP含量進行測定,測得其含量為6.64%,與核磁內標法測得的含量基本吻合,驗證了核磁內標法測定純度的可靠性。
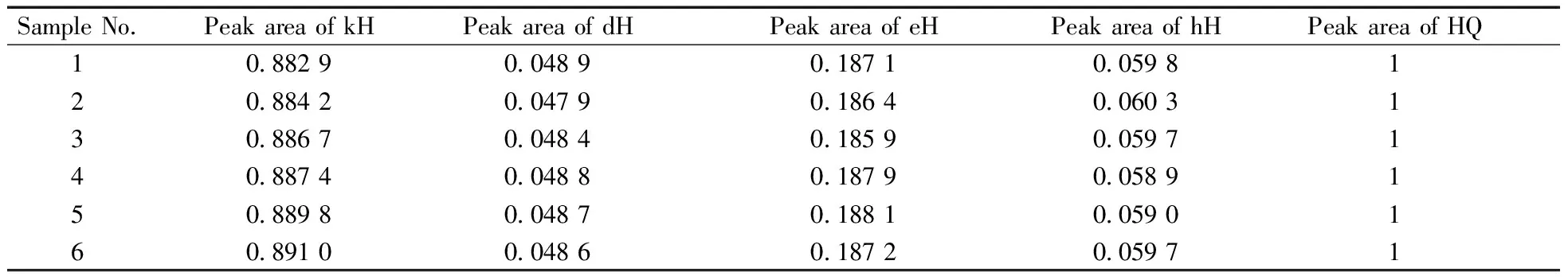
表1 N催化劑與對苯二酚積分面積的測定結果Table 1 The integral area of N-catalyst and HQ
此外,N催化劑制備時,在進行滴加TiCl4析出MgCl2、內給電子體和鈦負載處理兩個步驟之前的鄰苯二甲酸酐溶解液中,各活性組分的含量及濃度也可通過“1.4”方法計算得到。經計算,此時鄰苯二甲酸酐溶解液體系中的活性組分為TBP、PA-DCP-MgCl和DCP-MgCl,以HQ為內標物計算它們在體系中的濃度分別為0.408、0.058、0.188 mol/L。由之前的研究結果可知,十分微量的TBP在該體系中發生化學反應,而此時若以TBP為該體系內標物,體系中各活性組分的濃度分別為0.467、0.067、0.167 mol/L,較HQ為內標物時有明顯變化,說明在制備N催化劑時,作為助溶劑的TBP在體系中的化學反應量不可忽略。
3 結 論
優選了計算N催化劑體系活性組分的內標物質,以氘代甲醇為溶劑,經譜峰化學位移對比,選擇對苯二酚作為內標物質,以核磁內標法測定N催化劑樣品中活性組分的含量,溶劑峰和內標定量峰對樣品的定量峰無干擾,其結果的重復性和可靠性良好。該方法具有不需標準對照品、快速、高效等特點,在利用核磁技術研究N催化劑中活性組分的化學結構時可同時進行含量測定,為給電子體含量的表征提供了新的途徑,從而加深了對N催化劑優異聚合性能的理解,使N催化劑中活性組分的改進和性能的提高有了更多的可能性。
[1] Kashiwa N.J.Polym.Sci.PartA:Polym.Chem.,2004,42 (1): 1-8.
[2] Dong X F,Cui X P,Yang M,Liu B Y.Polym.Mater.Sci.Eng.(董小芳,崔曉鵬,楊敏,劉賓元.高分子材料科學與工程),2017,33 (3): 25-29.
[3] Ling Y T,Xia X Z,Liu Y X.Petrochem.Technol.(凌永泰,夏先知,劉月祥.石油化工),2017,46 (4): 422-426.
[4] Yin J,Zhao M J.Petrochem.Technol.(殷杰,趙梅君.石油化工),2014,44(1): 42-46.
[5] Yin J.Petrochem.Technol.(殷杰.石油化工),2017,46 (1): 124-129.
[6] Deng Z W,Li J,Xu M F,Liu P,Geng Z F.J.Instrum.Anal.(鄧志威,李璟,許美鳳,劉鵬,耿珠峰.分析測試學報),2012,31(9): 1081-1088.
[7] Deng X J,Li W B,Liu S N,Ding G S.Chin.J.Anal.Lab.(鄧小娟,李文斌,劉塞納,丁國生.分析試驗室),2017,36(9): 1032-1035.
[8] Zhang Y J,Liu X P.Chin.J.Magn.Reson.(張友杰,劉小鵬.波譜學雜志),2007,24(3): 289-295.
[9] Zhang W,Xu B.Chem.Anal.Meter.(張偉,徐蓓.化學分析計量),2004,13(6): 39-41.
[10] Zhou Q L,Tan Z,Yan L A,Xu X D,Song W W.Petrochem.Technol.(周奇龍,譚忠,嚴立安,徐秀東,宋維瑋.石油化工),2010,39(9): 997-1000.
[11] Wang Q,Wang M T,Zhang Z Q.J.Instrum.Anal.(王強,汪茂田,張志權.分析測試學報),2003,22(6): 101-103.
[12] Sun J X,Zhang Z X.Chin.J.Pharm.Anal.(孫靜霞,張正行.藥物分析雜志),2005,25(1): 117-122.
[13] Hu M,Hu C Q,Liu W Y.Chin.J.Anal.Chem.(胡敏,胡昌勤,劉文英.分析化學),2004,32(4): 451-455.
[14] Yin J,Zhao M J,Wang H.Chin.J.Anal.Lab.(殷杰,趙梅君,王紅.分析試驗室),2014,33(8): 972-974.
[15] Liu K,Wang M C,Xu M,Zhang L H,Zhang G,Chen Z Q.J.Instrum.Anal.(劉可,王民昌,徐敏,張麗涵,張皋,陳智群.分析測試學報),2017,36(3): 414-417.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
