microRNA靶基因預測算法的研究與發展
2018-03-02 08:04:07郭茂祖武雪劍
智能計算機與應用 2018年1期
程 爽, 郭茂祖, 武雪劍
(哈爾濱工業大學 計算機科學與技術學院, 哈爾濱 150001)
引言
MicroRNAs(miRNAs)是一類長度約19—22nt的非編碼單鏈RNA分子,是后轉錄表達過程的重要角色[1]。miRNAs綁定在RNA沉默復合物(RISC)上,引導該復合物與靶標mRNAs上的特殊位點配對,控制mRNA降解或抑制蛋白質翻譯。目前的生物實驗已證實miRNA廣泛存在生物體中,一個miRNA能調控超過100種mRNA。同時,人體中有超過60%的蛋白編碼基因的mRNA 3’UTR區域包含有miRNA結合位點。研究發現,miRNA優勢調控了大量的細胞進程,包括細胞增殖、新陳代謝和死亡,這就充分說明了miRNA在動物體內基因調控過程中的現實重要性[1]。然而到目前為止,miRNA與基因的靶向規則依然未能收獲最高研究成果,為了揭示miRNA的功能,識別miRNA靶向機制以及靶標基因即已成為當前亟待解決的問題。具體分析可知,miRNA的研究一直以來就十分活躍,與miRNAs相關的文獻發表數量也逐年攀升,值得一提的是,miRNAs靶向基因的識別方法在2003年以后即獲成熟實現與高效提出。比如一些文章集中研究生物學原理及檢驗方法[2-3],生物實驗技術和計算預測算法[4-6]。近期,大量文獻又深度探討了miRNA-mRNA的相互作用機制[7-10]。
本文首先介紹最近發表的miRNA靶向基因預測算法,總結miRNA目標預測的研究進展。miRNA靶向基因預測算法主要分為2類:ab initio計算方法和機器學習方法,依據這一分類,本文綜述了具有代表性的方法,最后,討論目前的挑戰和未來方向。
1 實驗數據
目前用于存放和共享miRNA-mRNA交互實驗數據的數據庫已經被開發、并成功創立。TarBase[11]數據庫始建于2006年,經過不斷的發展,目前版本為TarBase v7,其中收錄了近50萬的miRNA-gene的交互數據。TarBase提供了關于每個交互的更多細節,并且可以基于使用的實驗技術,調控類型(上調,下調與位置)與交互類型。miRecords[12]發布于2008年,迄今為止,該數據庫中收錄了由644miRNAs和1 901個靶基因組成的大約2 705條記錄。miRTarBase[13]收集miRNA-mRNA交互并且將miRNA-mRNA相互作用分為4類,包括功能性的、弱功能性的(非直接實驗支持)、非功能性的和弱非功能性的,依賴于所用實驗技術的力量和交互的類型(積極的和消極的)。這種分類方法適用于關聯研究。starBase[14]重點選取了CLIP-seq數據,該類型的數據包含轉錄組范圍規模上的mRNA-miRNA-Argonaute復合物的交互位點。PMRD[15]是一個關于植物microRNA數據庫,包含了miRNA序列、miRNA的靶基因、二級結構、表達譜和基因組信息等。miRNAMap[16]數據庫則動態集成了實驗證明的人類的、小鼠、大鼠以及其他多細胞動物的miRNA靶基因。
2 miRNA靶向特征
miRNA靶基因識別的關鍵步驟是有效特征的選擇。時下,經過研究發現了大量的特征,但是目前只有一些通用miRNA-mRNA靶向規則獲得學界認可,并相繼應用在各種靶基因預測算法中。考慮miRNA和mRNA交互種子區域的重要性,即使得常規分類多呈現為種子區域和非種子區域,其中,種子區域的miRNA序列則尤其重要[17],大量實驗又進一步發現了種子區域的強互補性。同樣,非種子區域在雙重交互和加強吸引力方面也已成為一個重要角色,發揮出色作用[18]。其次,描述相互作用強度的最小自由能也展現了miRNA-mRNA普遍具有的特征[19]。第三,miRNA與靶基因的結合位點的序列具有明顯保守性,甚至是跨物種的保守性[20]。
3 計算方法
自2003年以來,miRNA靶向基因預測算法的研究已經歷了十余年的發展歷程。當前基于結構特征的預測算法可劃分為2類:ab initio計算方法和機器學習方法。總地來說,最初提出的算法都是屬于ab initio類型,這些算法均是基于實驗得出的結構特征來指引預測[20-25]。另一方面,機器學習方法[26-34]則是基于實驗訓練集的相似度來識別候選靶向目標,部分原因可歸結為:機器學習算法本就是在實驗支持的靶向交互數據數量顯著增多時才應運而生的。下面即對上述2類中的代表性研究成果進行逐一闡述及對比剖析。
3.1 ab initio方法
(1)miRanda[20]。適用范圍廣泛,不受物種限制。該算法考慮序列匹配,miRNA-mRNA雙鏈的熱穩定性以及靶位點的跨物種保守性。其中,為了體現miRNA3’端、5’端和靶基因作用過程中的不對稱性,該軟件提出了scale參數。同時強調miRNA第2~4位堿基和靶基因精確互補,第3~12位堿基和靶基因錯配不得多于5個等特征。
(2)TargetScan[17]算法。對靶基因跨物種保守性和miRNA-mRNA雙鏈二聚體熱力學特征做出限制。需要至少6nt的種子互補并且考慮種子類型。TargetScan發布的最新版本添加了一些額外的約束條件,例如種子配對穩定性和目標位點豐富性。
(3)PicTar[23]算法。關于種子區域制定了嚴格的要求,強調miRNA-mRNA二聚體結合能在靶基因翻譯抑制中的關鍵作用。同時也要一并考慮基于最小自由能的miRNA-mRNA雙鏈穩定性。一旦位點匹配,可采用隱馬爾科夫模型給候選靶位點評分。
(4)RNA22[35]算法。是基于模式的發掘策略來識別候選目標。該算法強調miRNA-mRNA二聚體的互補匹配特性和自由能,但不考慮靶基因的跨物種保守性。首先,馬爾科夫鏈用于模式發現,識別與miRNA匹配的目標區域。其次,基于用戶設定的參數(配對堿基極小值,未配對堿基極大值,允許自由能的極大值)來選擇候選靶向區域。
(5)RNAhybrid[36]算法。基于miRNA-mRNA二聚體二級結構的最小自由能這一特征,不僅考慮靶向結合位點的能量,也考慮miRNA-mRNA雙鏈的能量,但不再關注靶基因的跨物種保守性。RNAhybrid允許用戶自定義自由能閾值及p值,也允許用戶設置雜交位點的偏好等特征。
(6)PITA[37]算法。不僅考慮特定二聚體互補匹配信息,還引入了mRNA位點的可接近性。可接近性表示了整個二聚體的最小自由能與互補匹配區域的原始能量ΔΔG之間的區別。用戶能夠強加不同的限制來減少候選合成集合(最小種子序列長度,G∶U錯配與未配對個數)。
(7)EiMMo[25]算法。使用貝葉斯方法來給候選靶向位點打分,研究直系同源物種之間靶向位點的演化,并推斷功能性靶向位點的系統分布。
(8)DIANA[38]算法。基于以下2點來判別miRNA靶基因:
① miRNA和靶基因間的高親和力,主要通過結合能來衡量;
② 影響miRNA和靶基因所形成二聚體莖環結構環部位置和環大小的miRNA相關蛋白可能指導miRNA和靶基因的相互作用。同時,衡量每個基因時,不僅考慮保守位點,也包括不保守的位點。
在此,研究可得如上各類方法的主題網址及實用預測結果的對照比較,具體見表1。通過上述內容分析可知,ab initio算法的不足是預測的結果假陽性頗高。其有效的技術策略是,通過上述內容分析可知,ab initio算法使用嚴格的限制條件來減少假陽性預測結果的數量。然而,過濾也會使一些真正的靶基因發生丟棄。比如PicTar,TargetScan和DIANA,為了獲取可觀的正確度(約50%),這些算法具有顯著妥協的敏感度(約10%)。而機器學習算法是實驗支持的反例數量偏低(反例通常不會公開發布并且不會記錄在數據庫中),導致正例和反例數量未臻均衡,影響分類模型的預測準確度。因此,接下來在分述機器學習算法的同時,也將全面拓展式地概述了這些方法所用的數據。
3.2 機器學習方法
(1)TargetBoost[26]算法。采用GPboost模型,考慮miRNA-mRNA二聚體的序列互補配對、熱力學穩定性、跨物種保守性等特征,預測線蟲和果蠅miRNA的靶基因。用于訓練的反例數據集包含300個隨機生成的序列,而正例數據集包含36個實驗驗證的miRNA-mRNA靶向關系。
(2)miTarget[27]算法。采用支持向量機方法,使用徑向基函數,預測目標候選。其中考慮miRNA-mRNA二聚體的結構特征、熱力學特征及堿基互補匹配等特征,但并未考慮靶基因的跨物種保守性。用于訓練支持向量機的反例數據集包括83個實驗驗證miRNA-mRNA靶向關系和163個通過實驗數據推理得出的miRNA-mRNA靶向關系,正例數據集包括152個miRNA-mRNA靶向關系。
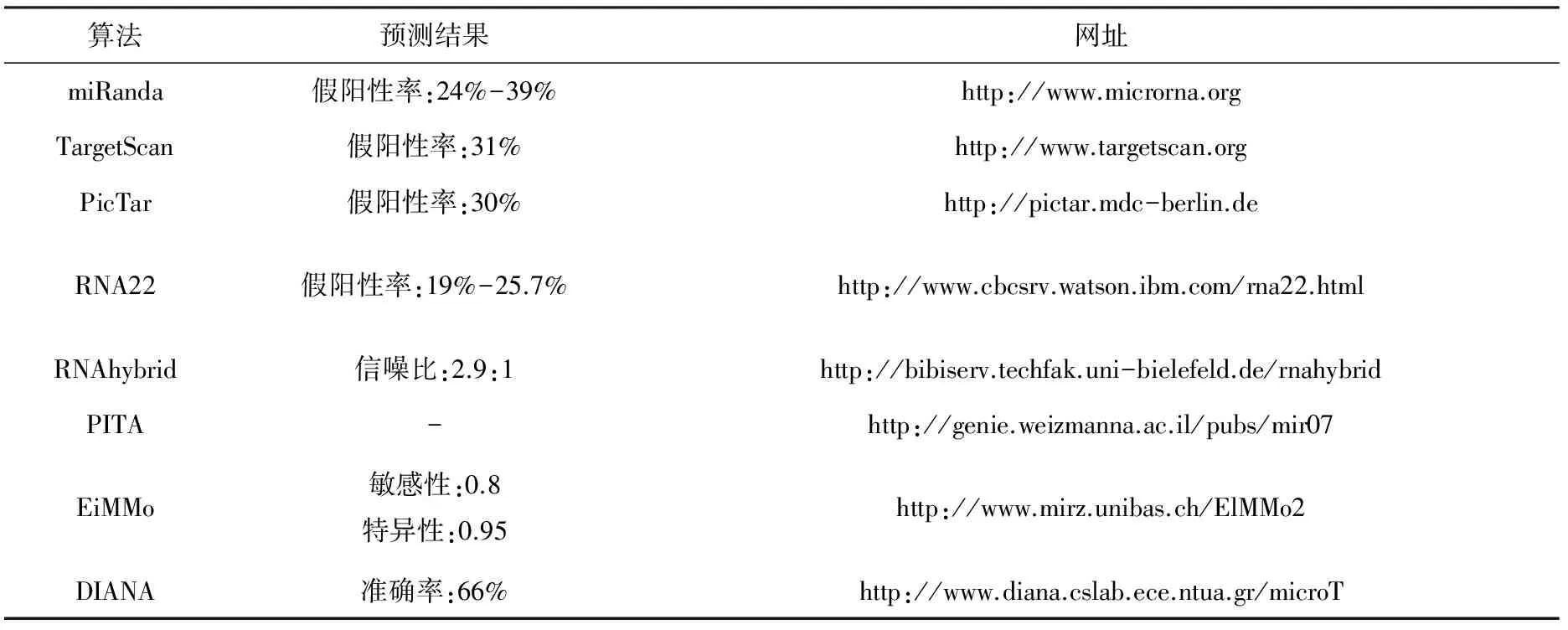
表1 預測miRNA靶基因的代表性ab initio方法Tab. 1 ab initio methods for miRNA target prediction
(3)Ensemble[28]算法。首先利用miRanda從miRNA-mRNA雙鏈中提取特征,之后采用多核SVM進行預測。正例數據集包含48個實驗驗證的miRNA-mRNA交互,反例數據集包含16個實驗驗證的反例miRNA-mRNA交互。
(4)NBmiRTar[29]算法。首先利用miRanda基于自由能和互補匹配的過濾條件篩選候選靶基因,之后利用樸素貝葉斯分類器計算每個候選靶基因的得分。反例數據集由38個實驗驗證的和133 316個人工選擇的候選靶位點組成,正例數據集由225個實驗驗證的miRNA-mRNA交互組成。
(5)MiRTif[31]算法。首先綜合了miRanda,PicTar和TargetScan這3種預測方法得到的各種特征得分。然后使用支持向量機方法預測候選靶基因,核函數采用徑向基函數。正例數據集包含195個實驗驗證的靶向交互,反例數據集包含了21個實驗驗證的和17個假定的靶向交互。
(6)TargetMiner[32]算法。首先基于種子區域互補匹配的特性,選取合適的靶向位點,并得出相應的特征得分。然后以此為依據,使用支持向量機模型預測靶基因,核函數采用徑向基函數。正例數據集包含764個靶向交互,反例數據集包含59個實驗驗證的和289個假定的靶向交互組成。
(7)MTar[33]算法。選擇了3類區域的靶向位點,計算相應區域miRNA-mRNA交互的特征得分(僅5’種子區域,5’種子區域占主要低位和以3’區域為主),然后使用人工神經網絡預測候選靶基因。正例數據集包含340個miRNA-mRNA靶向交互,反例數據集包含400個隨機的miRNA-mRNA靶向交互。
(8)TargetSpy[34]算法。首先生成候選靶向結合位點,對候選靶向位點排序。并計算候選靶向位點的堿基組成,結構特征以及堿基匹配互補等特征得分。正例集包含3 872個樣本,反例集合包含4 540個樣本。
(9)miREE[39]算法。首先利用遺傳算法來生成一個序列集合,之后利用支持向量機模型,采用徑向基函數,來預測候選靶基因。正例數據集包含324個靶向交互,反例數據集包含351個靶向交互。
(10)Target-align[40]。是一個基于Smith-Waterman的miRNA靶基因預測軟件。為了得到局部最優排列,Target-align依據堿基互補匹配程度構建了得分矩陣,并采用動態規劃算法預測miRNA靶基因。
(11)miRTDL[41]算法。考慮了miRNA-mRNA之間的互補匹配性、可接近性、保守性等特征。實驗選擇了1 297個實驗驗證的正例樣本,309個實驗驗證的反例樣本,由于反例樣本數量遠遠少于正例樣本數量,因此,該算法首先利用約束松弛方法構建了均衡的正、反例數據集,之后采用深度學習模型,預測miRNA的靶基因。
至此,各種算法關聯的主題網址及機器學習方法的整體展現可見表2。

表2 預測miRNA靶基因的代表性機器學習方法Tab. 2 Machine learning methods for miRNA target prediction
綜上可知,這些方法大多利用miRNA和基因的二級結構層面的序列、能量等特征,選用計算方法識別miRNA的靶基因。近年來,還有一些研究者利用miRNA表達譜數據或生物通路,以求通過表達值或miRNA在通路中的變化,來研究miRNA與基因的靶向關系。
3.3 其它方法
(1)MiRonTop[42]軟件。利用DNA微陣列數據和高通量測序數據來識別特定生物過程中潛在的miRNA靶基因,并設計了一定在線服務。用戶通過這個軟件可以快速查詢由其它軟件預測得出的靶基因。同時,通過調查靶位點在3’非編碼區域的位置,該軟件能夠將候選靶基因通過富集分析得到的最終得分提供給用戶。
(2)miRTar[43]軟件。分析了各種通路情況來識別基因轉錄本上的miRNA靶位點,并依據靶基因所在的生物通路來闡釋miRNA的生物功能。這個軟件通過分析特定通路來識別感興趣的miRNA和基因之間的調控關系,進而闡釋生物通路中miRNA真實發揮的具體作用。
(3)mirSOM[44]軟件。利用自組織圖(self-organizing map)聚類方法,對3’非編碼區域的序列進行聚類,該方法不僅能識別種子區域完美匹配的靶位點,也能發現不完美匹配的靶位點。
(4)文獻[45]。分別將關聯模型(Pearson)、因果推理模型(IDA)和回歸模型(Lasso)應用到表達數據上,將得到的3類結果進行綜合集成分析,由此證明集成方法與使用任一種模型的運行設計相比,集成方法的實驗結果均占據了明確優勢。
(5)文獻[46]。通過研究表達譜數據,分析miRNA和基因在不同通路中的表達情況,利用統計分析來確定miRNA的靶向基因。結果證明有21個miRNA在重疊的通路中得到了探知發現。
4 結束語
時下,雖然已經推出了大量的數據庫和預測軟件來預測miRNA的靶基因,但是其中的大部分卻都具有較高的假陽性,因此,預測miRNA的靶基因仍是學界的挑戰性研究課題。當今研究已經引入了保守性和功能相似性來降低假陽性結果,但應該指出靶基因預測準確率也同樣存在著可觀的提升完善空間。比如,可以參考各種疾病通路,依據miRNA和基因在通路中的表達,來篩選候選靶基因。其次,隨著生物實驗的高端推進,會有越來越多的靶向規則將進入學界視野,這也將顯著提高miRNA靶基因預測的準確性。另外,隨著高通量技術的飛速發展,使得短時間內識別特定miRNA的靶基因終將成為現實可能。
[1] BARTEL D P. MicroRNAs: genomics, biogenesis, mechanism, and function[J]. Cell, 2004,116(2):281-297.
[2]CAI Y, YU X, HU S, et al. A brief review on the mechanisms of miRNA regulation[J]. Genomics Proteomics Bioinformatics, 2009,7(4):147-154.
[3]MENDES N D, FREITAS A T, SAGOT M F. Current tools for the identification of miRNA genes and their targets[J]. Nucleic Acids Res, 2009,37(8): 2419-2433.
[4]ALEXIOU P, MARAGKAKIS M, PAPADOPOULOS G L, et al. Lost in translation: An assessment and perspective for computational microRNA target identification[J]. Bioinformatics, 2009,25(23):3049-3055.
[5]WITKOS T M, KOSCIANSKA E, KRZYZOSIAK W J. Practical aspects of microRNA target prediction[J]. Curr Mol Med, 2011,11(2):93-109.
[6]MIN H, YOON S. Got target? Computational methods for microRNA target prediction and their extension[J]. Exp Mol Med, 2010,42(4): 233-244.
[7]OROM U A, LUND A H. Experimental identification of microRNA targets[J]. Gene, 2010,451(1/2): 1-5.
[8]JIN H, TUO W, LIAN H, et al. Strategies to identify microRNA targets: New advances[J]. N Biotechnol, 2010, 27(6):734-738.
[9]THOMSON D W, BRACKEN C P, GOODALL G J. Experimental strategies for microRNA target identification[J]. Nucleic Acids Res, 2011, 39(16): 6845-6853.
[10]REYES-HERRERA P H, FICARRA E. One decade of development and evolution of microRNA target prediction algorithms[J]. Genomics Proteomics Bioinformatics, 2012,10(5):254-263.
[11]VLACHOS I S, PARASKEVOPOULOU M D, KARAGKOUNI D, et al. DIANA-TarBase v7. 0: Indexing more than half a million experimentally supported miRNA: mRNA interactions[J]. Nucleic acids research, 2015,43( D1):153-159.
[12]XIAO F, ZUO Z, CAI G, et al. miRecords: An integrated resource for microRNA-target interactions[J]. Nucleic Acids Res, 2009, 37(Database issue): 105-110.
[13]HSU S D, LIN F M, WU W Y, et al. miRTarBase: A database curates experimentally validated microRNA-target interactions[J]. Nucleic Acids Res, 2011,39(Database issue):163-169.
[14]YANG J H, LI J H, SHAO P, et al. starBase: A database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data[J]. Nucleic Acids Res, 2011,39(Database issue):202-209.
[15]ZHANG Z, YU J, LI D, et al. PMRD: Plant microRNA database[J]. Nucleic Acids Res, 2010,38(Database issue):806-813.
[16]HSU S D, CHU C H, TSOU A P, et al. miRNAMap 2.0: Genomic maps of microRNAs in metazoan genomes[J]. Nucleic acids research, 2008,36(suppl 1):165-169.
[17]LEWIS B P, BURGE C B, BARTEL D P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets[J]. Cell, 2005,120(1):15-20.
[18]BARTEL D P. MicroRNAs: Target recognition and regulatory functions[J]. Cell, 2009,136(2):215-233.
[19]MüCKSTEIN U, TAFER H, HACKERMüLLER J, et al. The-rmodynamics of RNA-RNA binding[J]. Bioinformatics, 2006,22(10):1177-1182.
[20]ENRIGHT A J, JOHN B, GAUL U, et al. MicroRNA targets in Drosophila[J]. Genome biology, 2004, 5(1):R1.
[21]FRIEDMAN R C, FARH K K H, BURGE C B, et al. Most mammalian mRNAs are conserved targets of microRNAs[J]. Genome research, 2009,19(1):92-105.
[22]GARCIA D M, BAEK D, SHIN C, et al. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs[J]. Nature structural & molecular biology, 2011,18(10): 1139-1146.
[23]LALL S, GRüN D, KREK A, et al. A genome-wide map of conserved microRNA targets in C. elegans[J]. Current biology, 2006,16(5):460-471.
[24]MIRADA K C, HUYNH T, TAY Y, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes[J]. Cell, 2006,126(6):1203-1217.
[25]GAIDATZIS D, VAN NIMWEGEN E, HAUSSER J, et al. Inference of miRNA targets using evolutionary conservation and pathway analysis[J]. BMC bioinformatics, 2007,8(1):69.
[26]SAETROM O, Jr SNOVE O, SAETROM P. Weighted sequence motifs as an improved seeding step in microRNA target prediction algorithms[J]. RNA, 2005,11(7):995-1003.
[27]KIM S K, NAM J W, RHEE J K, et al. miTarget: MicroRNA target gene prediction using a support vector machine[J]. BMC bioinformatics, 2006,7(1):411.
[28]YAN X, CHAO T, TU K, et al. Improving the prediction of human microRNA target genes by using ensemble algorithm[J]. FEBS letters, 2007,581(8):1587-1593.
[29]YOUSEF M, JUNG S, KOSSENKOV A V, et al. Naive Bayes for microRNA target predictions--machine learning for microRNA targets[J]. Bioinformatics, 2007,23(22):2987-2992.
[30]WANG X, NAQA I M E. Prediction of both conserved and nonconserved microRNA targets in animals[J]. Bioinformatics, 2008,24(3):325-332.
[31]YANG Y, WANG Y P, LI K B. MiRTif: A support vector machine-based microRNA target interaction filter[J]. BMC bioinformatics, 2008,9(Suppl 12): 4.
[32]BANDYOPADHYAY S, MITRA R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples[J]. Bioinformatics, 2009,25( 20): 2625-2631.
[33]CHANDRA V, GIRIJADEVI R, NAIR A S, et al. MTar: A computational microRNA target prediction architecture for human transcriptome[J]. BMC Bioinformatics, 2010,11( Suppl 1): 2.
[34]STURM M, HACKENBERG M, LANGENBERGER D, et al. TargetSpy: A supervised machine learning approach for microRNA target prediction[J]. BMC Bioinformatics, 2010,11: 292.
[35]MIRANDA K C, HUYNH T, TAY Y, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes[J]. Cell, 2006,126( 6):1203-1217.
[36]KRüGER J, REHMSMEIER M. RNAhybrid: MicroRNA target prediction easy, fast and flexible[J]. Nucleic acids research, 2006,34(suppl 2):451-454.
[37]KERTESZ M, IOVINO N, UNNERSTALL U, et al. The role of site accessibility in microRNA target recognition[J]. Nat Genet, 2007,39(10):1278-1284.
[38]MARAGKAKIS M, RECZKO M, SIMOSSIS V A, et al. DIANA-microT web server: Elucidating MicroRNA functions through target prediction[J]. Nucleic Acids Res, 2009,37(Web Server issue):273-276.
[39]REYES-HERRERA P H, FICARRA E, ACQUAVIVA A, et al. miREE: miRNA recognition elements ensemble[J]. BMC bioinformatics, 2011,12(1):454.
[40]XIE F, ZHANG B. Target-align: A tool for plant microRNA target identification[J]. Bioinformatics, 2010,26 (23):3002-3003.
[41]CHENG S, GUO M, WANG C, et al. MiRTDL: A deep learning approach for miRNA target prediction[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2015, 36(36):1.
[42]Le BRIGAND K, ROBBE-SERMESANT K, MARI B, et al. MiRonTop: Mining microRNAs targets across large scale gene expression studies [J]. Bioinformatics, 2010, 26( 24):3131-3132.
[43]HSU J B, CHIU C M, HSU S D, et al. miRTar: An integrated system for identifying miRNA-target interactions in human[J]. BMC Bioinformatics, 2011,12:300.
[44]HEIKKINEN L, KOLEHMAINEN M, WONG G. Prediction of microRNA targets in Caenorhabditis elegans using a self-organizing map[J]. Bioinformatics, 2011,27( 9):1247-1254.
[45]LE T D, ZHANG J, LIU L, et al. Ensemble methods for MiRNA target prediction from expression data[J]. PLoS One, 2015,10( 6): e0131627.
[46]XU P, ZHU Y, SUN B, et al. Colorectal cancer characterization and therapeutic target prediction based on microRNA expression profile[J]. Sci Rep, 2016,6: 20616.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
兒童故事畫報(2019年5期)2019-05-26 14:26:14
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
