HPLC-MS/MS法發現ssaX阻斷菌株中兩個尿苷肽類新化合物
2018-02-27 06:10:33江志波侍媛媛任衛聰范佳會雷璇李星星王麗非解云英洪斌
中國醫藥生物技術 2018年1期
關鍵詞:結構
江志波,侍媛媛,任衛聰,范佳會,雷璇,李星星,王麗非,解云英,洪斌
?
HPLC-MS/MS法發現阻斷菌株中兩個尿苷肽類新化合物
江志波*,侍媛媛*,任衛聰,范佳會,雷璇,李星星,王麗非,解云英,洪斌
100050 北京,中國醫學科學院北京協和醫學院醫藥生物技術研究所/衛計委抗生素生物工程重點實驗室
對基因阻斷菌株次級代謝產物中的 sansanmycin 類似物進行研究,發現新化合物。
對sp. SS 野生菌株及基因阻斷株的發酵液分別進行固相萃取,收集 60% 甲醇水洗脫液進行 HPLC-MS/MS 分析。通過與文獻報道的已知化合物數據相比較的方法,分析其中 sansanmycin 類化合物結構并利用 MS/MS 對其結構進行確證。
從基因阻斷株的發酵液中發現了 8 個 sansanmycin 類化合物,其中SS-MX-7 和 SS-MX-8 為新結構化合物。SS-MX-7 化學結構中假四肽肽鏈上的第一位和第四位氨基酸殘基均為酪氨酸取代,而 SS-MX-8 的這兩個位置均為苯丙氨酸。
基因阻斷株產生的兩個新 sansanmycin 類似物,進一步豐富了尿苷肽類化合物的結構多樣性,同時也為利用HPLC-MS/MS 方法在其他重組菌株中發現尿苷肽類新化合物奠定了基礎。
尿苷肽類化合物;sp. SS; Sansanmycins; HPLC-MS/MS
尿苷肽類抗生素(uridyl peptide antibiotics,UPAs)包括 sansanmycins(SS)、pacidamycins、napsamycins 和 mureidomycins 等[1],它們具有共同的母核結構:由一個特殊結構的四肽單元通過酰胺鍵與4',5'-烯胺-3'-脫氧尿苷(尿苷部分)相連接。該類化合物對銅綠假單胞菌()和結核分枝桿菌()具有生長抑制作用,并且對結核分枝桿菌耐藥菌株有效[1-2]。尿苷肽類化合物通過抑制細菌肽聚糖合成中一個關鍵酶(磷酸--乙酰胞壁酸五肽轉位酶,MraY)的活性,從而阻斷細菌細胞壁的生成,抑制細胞生長[3-5]。
Sansanmycins 是由鏈霉菌sp. SS所產生的一組新型尿苷肽類抗生素[6]。目前已發現 30 余種類似物,它們結構中都具有一個(,)-3-甲基-2,3-二氨基丁酰基[(2S, 3S)-3-methyl-2,3- diamino butyric acid,DABA]結構單元,DABA 的引入導致肽鏈發生了分支(圖 1):3-甲氨基與 AA1偶聯,組成了肽鏈的 N-端;而 2-氨基與 AA3以肽鍵相連進一步延長了肽鏈;DABA 通過其羧基與 4',5'-烯胺-3'-脫氧尿苷相連接使得結構在縱向上得到延伸。結構中的脲基的摻入使得肽鏈在進一步延伸的過程中再次發生了方向改變,肽鏈由 N-端(AA3)轉變為C-端(AA4)。
Sansanmycin A(SS-A,圖 1)是在鏈霉菌sp. SS 發現的第一個尿苷肽類化合物,組成其四肽的氨基酸單元除 DABA 外,AA1、AA3和 AA4分別為間位酪氨酸(-Tyr),甲硫氨酸(Met)和色氨酸(Trp)[6]。該化合物對銅綠假單胞菌(ATCC 10145)和結核分枝桿菌(H37Ra)均有生長抑制作用,最低抑菌濃度(MIC)分別為 12.5 和 10.0 μg/ml[6]。Sansanmycin 獨特的化學結構、臨床上尚未開發的作用靶點以及良好的抗菌活性吸引著我們進一步發掘其類似物,期望獲得成藥性更優的抗結核先導化合物。
已報道的 sansanmycin 類化合物結構差異主要在組成四肽的氨基酸種類上,前期的研究表明 N-末端(AA1)和 C-末端(AA4)多數為芳香類氨基酸取代,且 AA1以間位酪氨酸、酪氨酸、苯丙氨酸以及雙環結構(bicyclic a/b,如SS-N/O,圖 1)為主[5-8],而 AA4在絕大多數情況下為色氨酸,少數為酪氨酸或苯丙氨酸[2, 7-8]。Sansanmycin 四肽骨架中的第三位氨基酸(AA3)通常為脂肪族氨基酸,如甲硫氨酸(Met)、甲硫氨酸亞砜(MetSO)和亮氨酸(Leu)[6, 9],少數情況下為苯丙氨酸[10]。
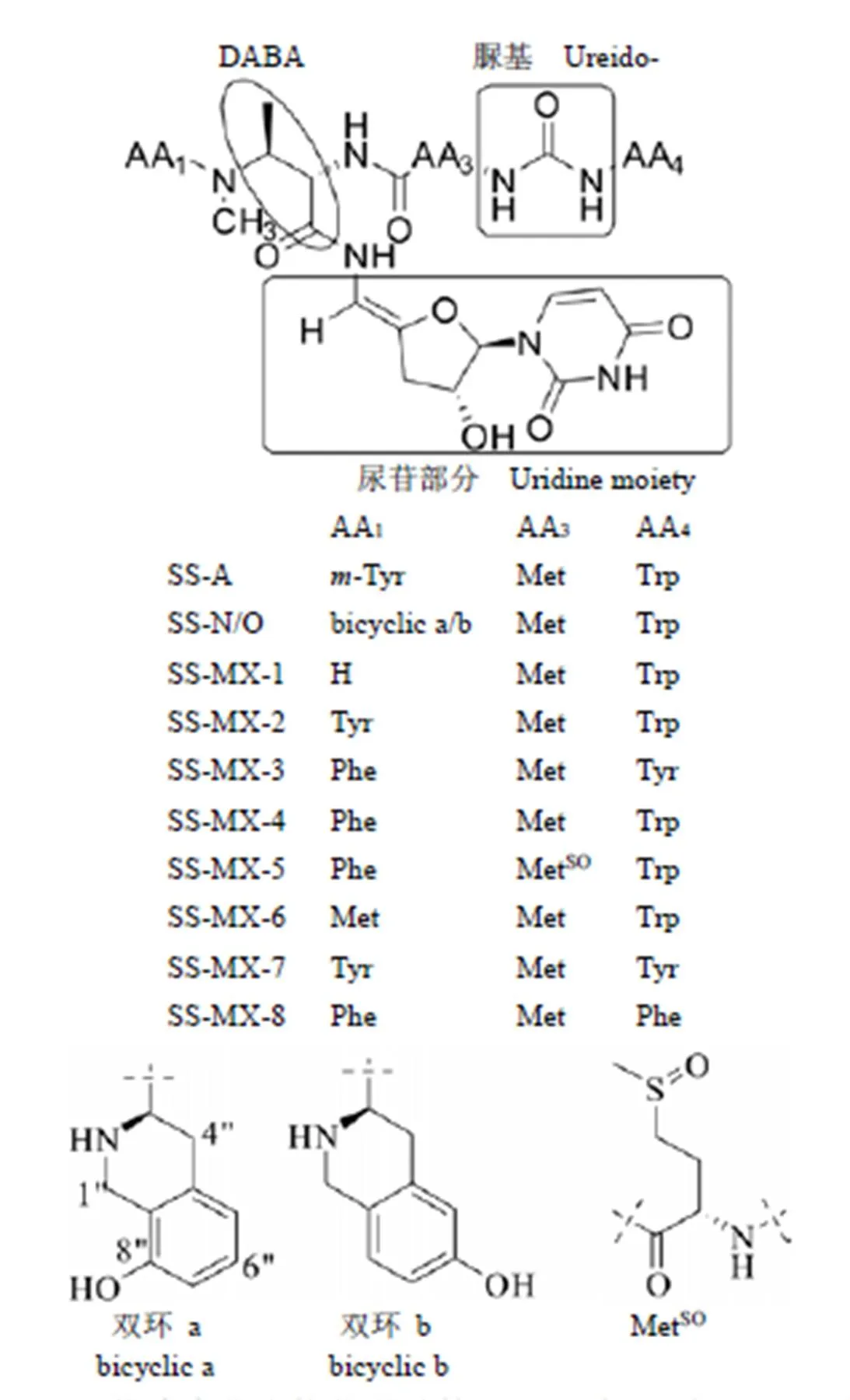
圖 1 尿苷肽類化合物化學結構(AA:氨基酸;Tyr:酪氨酸;Met:甲硫氨酸;Phe:苯丙氨酸;Trp:色氨酸;MetSO:甲硫氨酸亞砜)
Figure 1 The chemical structures of uridyl peptide antibiotics (AA: Amino acid; Tyr: Tyrosine; Met: Methionine; Phe: Phenylalanine; Trp: Tryptophan; MetSO: Methionine sulfoxide)
本課題組首次報道了 sansanmycin 生物合成基因簇[11],其包含幾個高度分散的非核糖體肽合成酶(nonribosomal peptide synthetases,NRPS)負責催化合成結構中的四肽單元。另外,還包括一些前體合成酶基因,如-Tyr 合成酶[12]基因(編碼苯丙氨酸-3-羥化酶)和DABA 合成酶基因等。前期,為了研究相關基因功能,我們構建了同框缺失菌株[2]。通過對基因阻斷菌株的代謝產物的分析發現,在阻斷后,不再產生含有-Tyr 的尿苷肽類化合物,從而驗證了的功能,在體內證明其為-Tyr 合成酶基因,負責合成-Tyr。與此同時,在此研究過程中發現了 6 個 sansanmycin 新類似物(SS-MX-1 ~SS-MX-6),說明負責加載 AA1的 NRPS(SsaU/W)具有一定的底物寬泛性;進一步通過底物飼喂的方法,以基因阻斷株為宿主,利用突變生物合成獲得了 20 余個新結構的 sansanmycin 類似物[2]。
為了進一步考察基因阻斷對 sansanmycin類化合物生物合成的影響,本文借助于HPLC-MS/MS 技術,對基因阻斷株和野生菌株發酵液粗提物進行了系統對比分析,比較基因阻斷株與野生菌株的代謝產物差異,在基因阻斷株的發酵液中發現了 8 個sansanmycin 類似物,通過與已知 sansanmycin 化合物的 MS/MS 譜進行比較解析了所有化合物結構,其中兩個為首次報道的新化合物(SS-MX-7 和 SS-MX-8)。
1 材料與方法
1.1 材料
1.1.1 菌株 鏈霉菌sp. SS(CPCC 200442)為中國藥學微生物菌種保藏管理中心提供;基因阻斷菌株構建方法見文獻[2]。
1.1.2 色譜試劑和耗材 色譜甲醇購自上海泰坦公司;色譜純水為屈臣氏蒸餾水經Millipore 純化儀純化而得;甲酸為化學級,購自北京化工廠;固相萃取柱(Sep-Pack C18)購自美國 Waters 公司;色譜柱(Capcell Pak C18 AQ,4.6 mm × 250 mm,5.0 μm)購自日本資生堂公司。
1.1.3 儀器 液相色譜儀Agilent 1100 和離子阱質譜儀 Agilent 5630 均為美國安捷倫公司產品。
1.2 方法
1.2.1 菌株活化、培養與發酵 各菌株接種于 100 ml 種子培養基,于 28 ℃、200 r/min 下振蕩培養 48 h 后,轉接到發酵培養基,相同條件下繼續培養 5 d,得發酵液。培養基配方參見文獻[2]。
1.2.2 樣品前處理 發酵液經 3000 r/min 離心 30 min,除去菌體和不溶物。取10 ml 上清液進行固相萃取,60% 甲醇水洗脫液經 0.22 μm 濾膜過濾后,用于HPLC-MS/MS 檢測。
1.2.3 HPLC-MS/MS 分析 液相色譜采用兩相梯度洗脫的方法,其中 A 相為甲醇,B相為 0.1%(V/V)甲酸水溶液。A 相在 70 min 內由 10%(V/V)逐漸遞增至 90 %(V/V),流速為 0.8 ml/min,洗脫液經過色譜柱后,使用三通等分至二極管陣列檢測器(diode array detector,DAD)和質譜檢測器中。檢測波長為 254 nm,柱溫箱設定為 25 ℃,進樣體積為 20 μl。
質譜檢測條件:噴霧電壓:4.5V;載氣:氮氣,流速10 L/min;離子源溫度:325 ℃;碰撞裂解氣體:高純氦氣(He);一級質譜(1D MS)使用全掃描模式,范圍為300 ~ 1200;二級質譜(MS/MS)采用自動采集模式,單次選擇兩個豐度高于 104分子離子進行采集;碰撞能量:20 kV;所有數據使用儀器自帶軟件 Data Analysis, version 3.3 進行處理。
2 結果
2.1 野生菌株主產物的結構分析
本實驗中野生菌株發酵液中顯示有 3 個豐度較高的保留時間分別為 37.4(峰 1)、38.1(峰 2)和 39.5(峰 3)min 色譜峰(圖 2A),其中峰 1 和3 分子離子均為876[M+H]+,峰 2 對應分子離子為864[M+H]+。峰 2 的二級質譜(圖 2B)給出特征碎片離子660(M-AA4),641(M-尿苷)和 701(M-AA1)與文獻[6]報道的主產物 SS-A 相吻合。
峰 1 和 3 給出的二級質譜很相似(圖 2C 和 2D),提示兩者可能為同分異構體。仔細比較這兩個峰和已知化合物 SS-N 和 SS-O[6]的二級質譜,發現它們并無差別。提示峰 1 和 3 應該為 SS-N 和(或)SS-O。由于 SS-N 和 SS-O 在結構上差別僅為 AA1上的羥基位置(SS-N 的羥基位于 C-6'',而 SS-O 的羥基連接在 C-8''),因此,兩者無法通過二級質譜的方法區分。
SS-N/O 與 SS-A 化學結構上差別僅為一個 AA1結構上的亞甲基,當 SS-A 溶解在甲醇水(1%,V/V)溶液中(2.0 mg/ml)靜置 48 min 后,HPLC 檢測結果顯示 SS-A 消失,在保留時間分別為 16.3 和 19.8 min 出現兩個峰(圖 3A 和 3B)質譜數據(ESI-MS,圖 3C)顯示這兩組峰對應的化合物的分子量876[M+H]+(圖 3C)與SS-N/O相同。分別制備兩組峰對應的化合物并進行高分辨二級質譜分析(圖 3D),結果顯示兩化合物的二級質譜及高分辨質譜數據與文獻報道 SS-N/O[10]完全一致,提示 SS-N/O 可能來源于 SS-A。
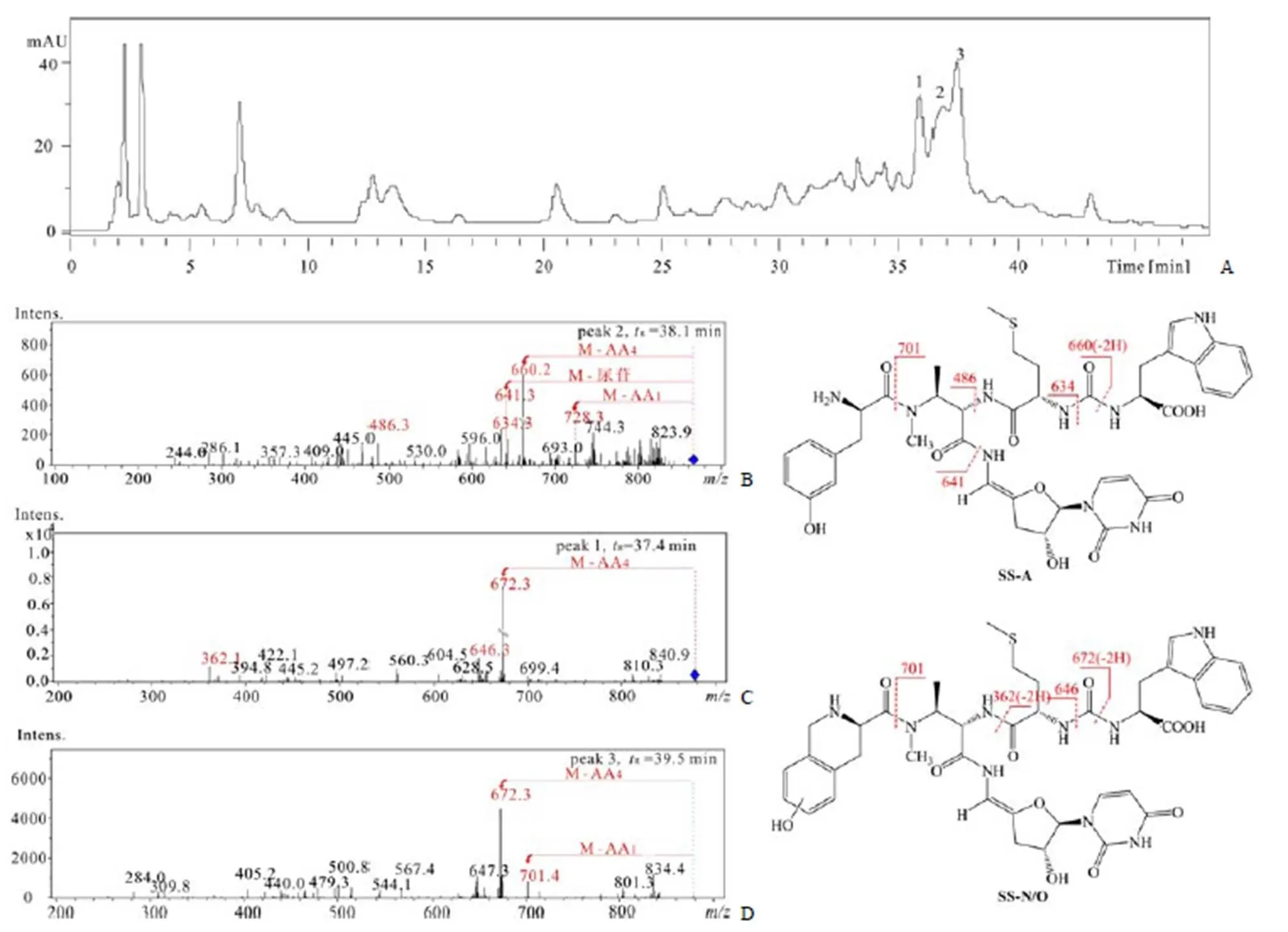
圖 2 野生菌株發酵液的HPLC-DAD 色譜圖(A)、峰 2(B)、峰 1(C)和峰3(D)的ESI-MS/MS 圖及化合物SS-A 和SS-N/O 裂解圖(紅色線為輔助線;碎片離子和丟失方式用紅色字體表示)
Figure 2 TheHPLC-DAD spectrum for the fermentation broth of wild type strain, and the ESI-MS/MS spectra of peak 2 (B), peak 1 (C) and peak 3 (D) and the fragmentation pathway of SS-A and SS-N/O (The red lines are the auxiliary lines, while the fragment ions and their lost way are in red font)
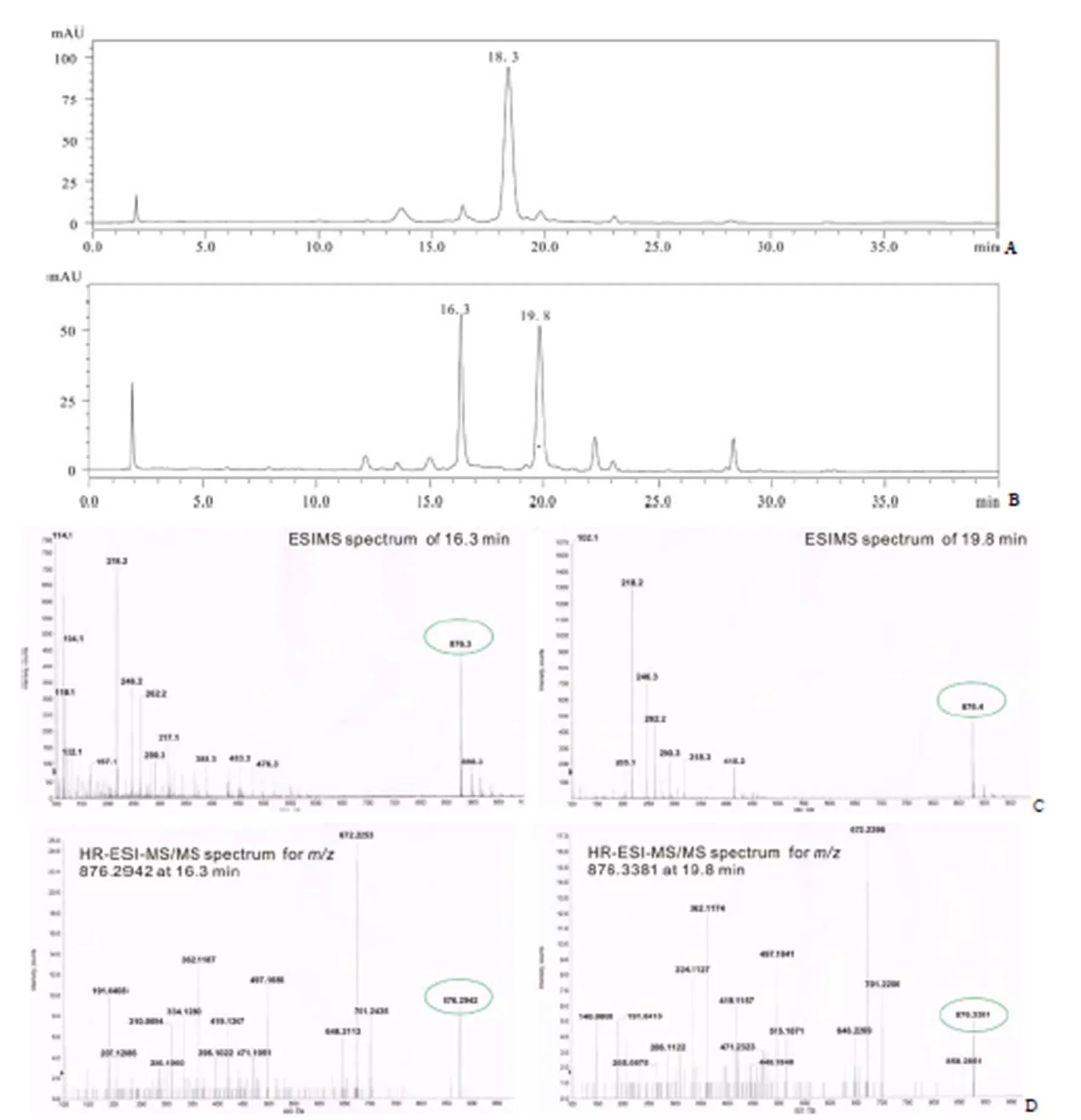
圖 3 化合物SS-A 與甲醇反應產物HPLC、ESI-MS 和HR-ESI-MS/MS 圖(A:反應0 h 的HPLC 圖;B:反應48 h 的HPLC 圖;C:圖B 中保留時間為16.3 和19.8 min 的ESI-MS 圖;D:圖B 中保留時間為16.3 和19.8 min 對應分子量分別為m/z 876.2942 和876.3381的HR-ESI-MS/MS 圖。綠色橢圓框為兩組化合物的對應分子離子峰)
Figure 3 The result for the reaction of SS-A in methanol aqueous solution (A: HPLC spectra for the reaction mixture at 0 h; B: HPLC spectra for the reaction mixture at 48 h; C: ESI-MS spectra at the peaks of 16.3 and 19.8 min in B respectively; D: HR-ESI-MS/MS for the molecular ions of876.2942 and 876.3381 in peaks of 16.3 and 19.8 min in B, respectively. The molecular ions were signed in green ovals)
2.2 ssaX基因阻斷株和野生菌株產物比較分析
HPLC 結果顯示(圖 4A),在 254 nm 下,野生菌株發酵液次級代謝產物較基因阻斷株豐富,特別是在 30 ~ 50 min,野生菌株發酵液峰強度強于基因阻斷株。保留時間為R37.4 ~ 39.5 min(圖 4A-1)的主成分 SS-A 和 SS-N/O 在基因阻斷株中完全消失,說明基因缺失對主成分的產生具有很大影響。
質譜基峰圖(base peak chromatography,BPC)是在質譜檢測中將每個時間點最強的離子連續描繪得到的圖譜,相比于總離子圖而言更加清晰簡潔。本實驗的質譜結果與HPLC-DAD 結果類似(圖 4B),從BPC 中來看,主峰 sansanmycin A(864[M+H]+;AA1為-Tyr)和 sansanmycin N/O(876[M+H]+;AA1為 bicyclic a/b)在基因阻斷株中完全消失,進一步說明基因的缺失影響了結構中含有-Tyr 片段的化合物的產生。
2.3 ssaX 基因阻斷菌株的液質結果分析
質譜BPC(圖 4B)顯示,基因阻斷株發酵液在 sansanmycins 區域(30 ~ 50 min)雖缺失了野生菌株中的三個主峰(SS-A 和 SS-N/O),但是還存在一些其他的離子峰,特別是保留時間為R34.0、35.1 和 39.0 min(圖 4B-2)處的三組峰,顯示具有和尿苷肽類化合物相似的紫外吸收,提示該菌株仍然具有產生 sansanmycin 類似物的能力。對圖 4B-2 出現的 sansanmycin 類似物的分子離子及其二級質譜進行分析,結果顯示,除前期研究中得到的 SS-MX-1 ~ SS-MX-6 外,還發現了兩個含有 4',5'-烯胺-3'-脫氧尿苷分子片段(225 D)的尿苷肽類化合物,分別命名為 SS-MX-7 與 SS-MX-8。
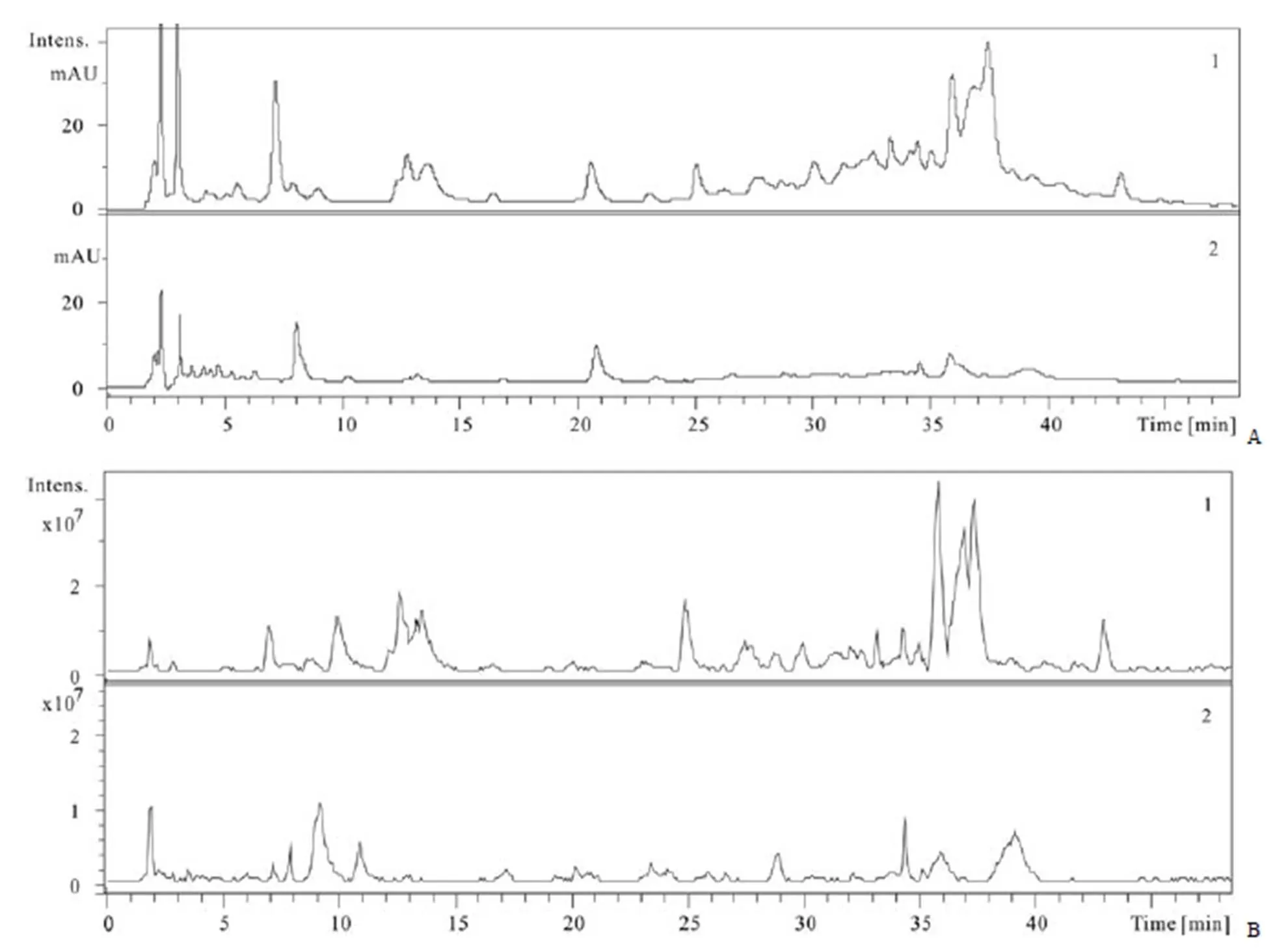
圖 4 HPLC-DAD(A)和質譜BPC(B)圖用于分析野生菌株和ssaX 基因阻斷株次級代謝產物的差異(A-1:野生菌株發酵液的HPLC 圖;A-2:ssaX 基因阻斷株發酵液的HPLC 圖;B-1:野生菌株發酵液的HPLC-MS/MS BPC 圖;B-2:ssaX 基因阻斷株發酵液的HPLC-MS/MS BPC 圖)
Figure 4 Comparison of the secondary metabolites of wild type andknockout strain by HPLC (A) and base peak chromatograph (BPC, B) (A-1: HPLC-DAD spectrum of wild type strain; A-2: HPLC-DAD spectrum ofknockout strain; B-1: BPC spectrum of wild type strain; B-2: BPC spectrum ofknockout strain)
2.4 ssaX 基因阻斷株中新化合物 SS-MX-7 和 SS-MX-8 結構推測
化合物SS-MX-7 保留時間為 30.5 min,在線紫外檢測顯示最大吸收波長分別為 215 和 262 nm,與文獻報道的化合物 SS-A 相似[5],推測其可能為 sansanmycin 類似物。對該化合物分子離子841[M + H]+的二級質譜進行分析(圖 5A),發現其二級質譜與 SS-MX-2[2]很相似,均具有碎片離子660[M+H]+,說明其除 C-末端氨基酸(AA4)之外的部分應該與 SS-MX-2 相同。這一推測得到了碎片離子616(M-AA4-脲基-2H)和碎片丟失 M-647 D(對應于碎片 AA1-NCH3)的支持。化合物尿苷部分經碎片丟失 M-616 D +2H(225 D)推測為4',5'-烯胺-3'-脫氧尿苷。由丟失碎片 M-660 D 推測化合物SS-MX-7 的AA4為酪氨酸(181 D)。
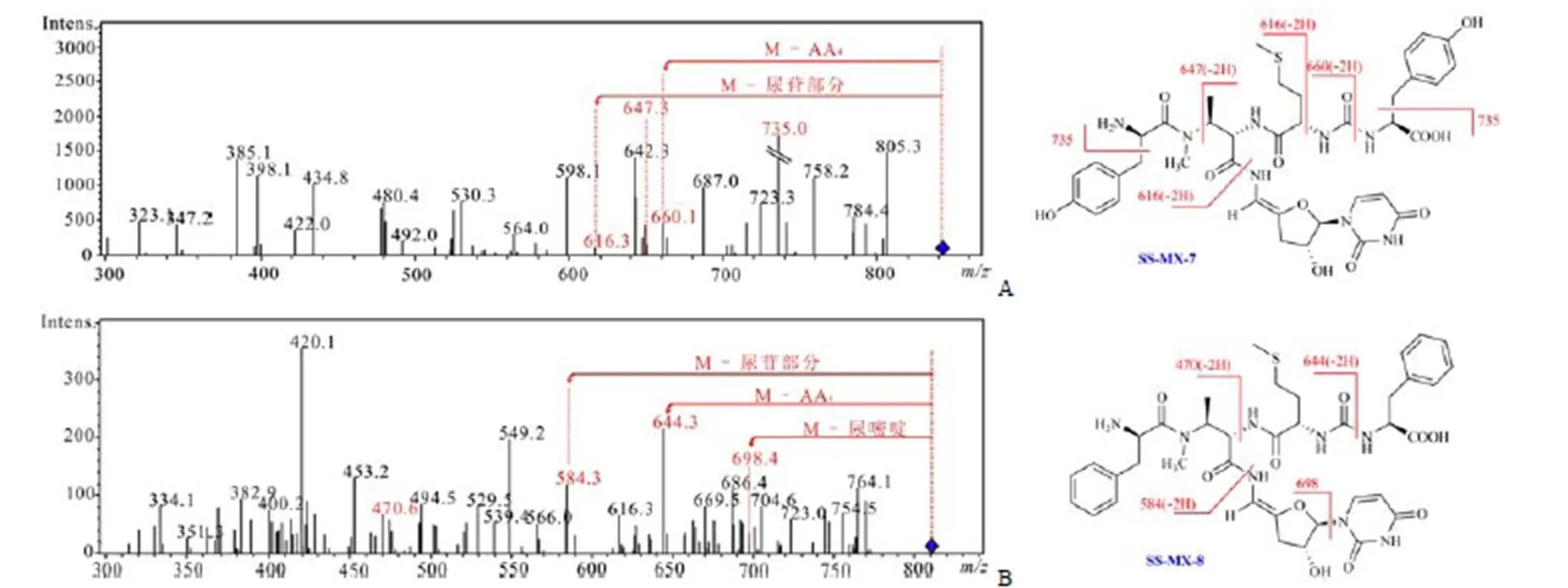
圖 5 化合物SS-MX-7 (A)和SS-MX-8(B)二級質譜分析(紅色線為輔助線;碎片離子和丟失方式用紅色字體表示)
Figure 5 Analysis of the MS/MS spectra of SS-MX-7 (A) and SS-MX-8 (B) (The red lines are the auxiliary lines, while the fragment ions and their lost way are in red font)
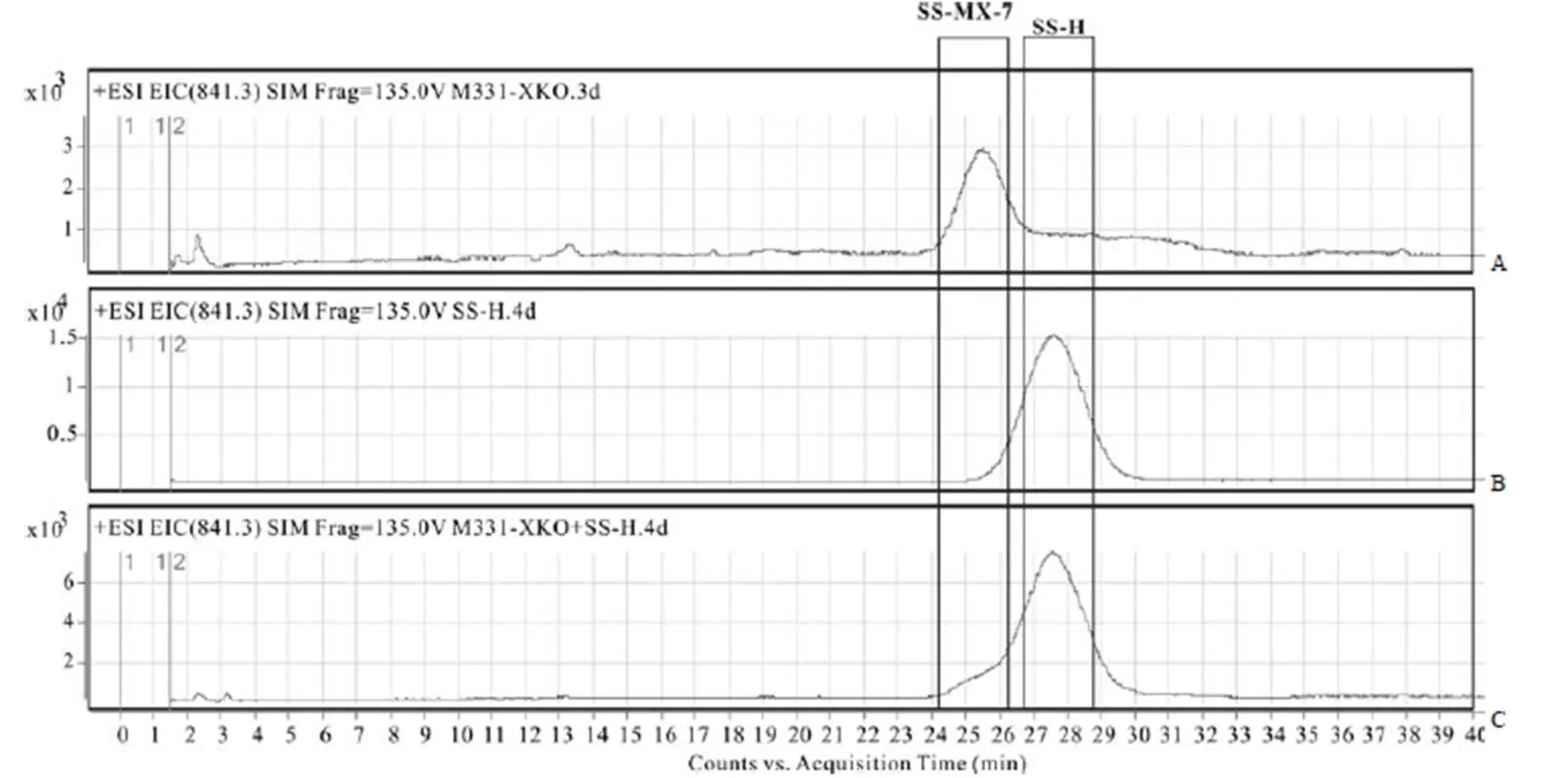
圖 6 單離子監測m/z 841.3 分析ssaX基因敲除菌株中的化合物SS-MX-7(A:ssaX 基因敲除菌株發酵液樣品;B:SS-H;C:在A 樣品中等體積加入SS-H)
Figure 6 Analysis of the compound SS-MX-7 in the fermentation broth ofknockout strain using the single ion monitor (SIM) of841.3 (A: Fermentation broth ofknockout strain; B: Purified SS-H; C: Equal volume of SS-H addition to sample A)
由于化合物 SS-MX-7 與已知化合物 SS-H[10](N-末端為-Tyr,C-末端為 Tyr)具有相同分子量,為了進一步驗證化合物 SS-MX-7 不是野生菌株中產生的 SS-H,借助于單離子監測(single ion monitor,SIM)方法對樣品阻斷菌株發酵液和 SS-H(4.6 mg/ml)同時進行 HPLC-MS 檢測(圖 6)。結果表明,阻斷菌株發酵液中的峰841.3(圖 6A)與 SS-H(圖 6B)保留時間不同,其中 SS-H 為 27.5 min,而阻斷菌株發酵液中峰841.3 為 25.0 min,提示 s阻斷菌株發酵液中 SS-MX-7 與 SS-H 結構不同。進一步將化合物 SS-H 等體積添加到阻斷菌株發酵液樣品中進行同樣實驗,根據保留時間差異(圖 6C)證明 SS-MX-7 與 SS-H 確實不是同一個化合物,SS-MX-7 為新化合物。
化合物SS-MX-8 保留時間為 38.0 min,分子離子為809[M + H]+。其紫外吸收同樣與文獻報道的化合物 SS-A 相似[5],提示SS-MX-8 可能也是尿苷肽類化合物。該化合物二級質譜(MS/MS,圖 5B)與文獻報道的SS-MX-4[2]有共同的特征離子644(M-AA4)和 470(M-AA3-AA4),推測該化合物除 C-末端之外的結構部分與 SS-MX-4 相同,即 AA1為苯丙氨酸(Phe),尿苷部分為4',5'-烯胺-3'-脫氧尿苷。由 M-644(165 D)可以推導出化合物SS-MX-8 的 C-末端同樣為苯丙氨酸。因此,化合物 SS-MX-8 是 N-末端和 C-末端均為苯丙氨酸的尿苷肽類新化合物。
3 討論
Sansanmycins 是由本所首次發現的一類新型尿苷肽類抗生素,對銅綠假單胞菌和結核分枝桿菌,尤其是臨床分離的多藥耐藥結核分枝桿菌有效,其主要通過特異性抑制細菌細胞壁合成過程肽聚糖合成酶 MraY 的活性來發揮作用。目前,臨床上還沒有針對該靶點的藥物,因此,該類化合物不易與臨床上在用的抗生素產生交叉耐藥,具有研發成為新型抗結核抗生素的潛力。
液相色譜(LC)和串聯質譜(MS/MS)聯用的方法是一種已經發展成熟的在線分析方法,其將液相的高效分離能力與質譜的高靈敏度、低檢出限以及多級質譜的結構定性分析能力有機結合在一起,已經成為發現新結構天然產物的有力幫手之一[13-17]。液質聯用技術除了具有快速分析復雜樣品的能力之外,還能對具有特定結構片段化合物進行聚類分析,能成簇地發現新結構化合物及其同系物[18]。除此之外,液質聯用在定量分析方面表現的優越性也已經成為藥物研究和新藥發現重要手段,特別是隨著串聯質譜技術發展和廣泛應用,借助于二級離子定量,可以定量分析同分異構體和具有微小結構差異的同系物。
本文中利用 HPLC-MS/MS 方法分析了野生菌株與基因阻斷株產生尿苷肽類化合物 sansanmycin 的差異,并成功地從基因阻斷株挖掘到之前沒有發現的兩個新結構 sansanmycin 類似物,進一步豐富了尿苷肽類化合物的結構多樣性。但在基因阻斷株中發現的sansanmycin 類似物產量都很低,尚難獲得足夠量進行生物活性的評價。目前 sansanmycin 的生物合成途徑已經基本解析,這些新結構的發現可以指導后續生物合成元件如特定底物選擇性的非核糖體肽合成酶(NRPS)的挖掘或定向進化,而后在合成生物學的指導下進行生物合成元件的替換與組合,有目的地合成新化合物,為篩選獲得成藥性更優的抗結核先導化合物提供物質基礎。
[1] Winn M, Goss RJ, Kimurab K, et al. Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat Prod Rep, 2010, 27(2):279-304.
[2] Shi Y, Jiang Z, Lei X, et al. Improving the N-terminal diversity of sansanmycin through mutasynthesis. Microb Cell Fact, 2016, 15:77.
[3] Lloyd AJ, Brandish PE, Gilbey AM, et al. Phospho-N-acetyl- muramyl-pentapeptide translocase from Escherichia coli: catalytic role of conserved aspartic acid residues. J Bacteriol, 2004, 186(6):1747- 1757.
[4] Al-Dabbagh B, Henry X, Ghachi ME, et al. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry, 2008, 47(34):8919- 8928.
[5] Hakulinen JK, Hering J, Br?ndén G, et al. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat Chem Biol, 2017, 13(3):265-267.
[6] Xie Y, Chen R, Si S, et al. A new nucleosidyl-peptide antibiotic, sansanmycin. J Antibiot (Tokyo), 2007, 60(2):158-161.
[7] Xie YY, Xu HZ, Yu Y, et al. Isolation, purification and structure determination of uridylpeptide antibiotic sansanmycin E. ChinJ Antibiot, 2009, 34(6):326-328. (in Chinese)解云英, 許鴻章, 俞瑩, 等. 尿苷肽類抗生素sansanmycin E的分離、純化和結構鑒定. 中國抗生素雜志, 2009, 34(6):326-328.
[8] Xie YY, Xu HZ, Yu Y, et al. Isolation, purification and structure determination of uridylpeptide antibiotic sansanmycin D with activity against Pseudomonas aeruginosa. Chin J Antibiot, 2009, 34(1):12-14, 44. (in Chinese)
解云英, 許鴻章, 俞瑩, 等. 尿苷肽類抗生素sansanmycin D的分離、純化和結構鑒定. 中國抗生素雜志, 2009, 34(1):12-14, 44.
[9] Xie Y, Xu H, Sun C, et al. Two novel nucleosidyl-peptide antibiotics: Sansanmycin F and G produced by Streptomyces sp. SS. J Antibiot (Tokyo), 2010, 63(3):143-146.
[10] Xie Y, Cai Q, Ren H, et al. NRPS substrate promiscuity leads to more potent antitubercular sansanmycin analogues. J Nat Prod, 2014, 77(7):1744-1748.
[11] Li Q, Wang L, Xie Y, et al. SsaA, a member of a novel class of transcriptional regulators, controls sansanmycin production in Streptomyces sp. strain SS through a feedback mechanism. J Bacteriol, 2013, 195(10):2232-2243.
[12] Zhang W, Ames BD, Walsh CT. Identification of phenylalanine 3-hydroxylase for meta-tyrosine biosynthesis. Biochemistry, 2011, 50(24):5401-5403.
[13] Lam KS. New aspects of natural products in drug discovery. Trends Microbiol, 2007, 15(6):279-289.
[14] Hol?apek M, Jirásko R, Lísa M. Recent developments in liquid chromatography-mass spectrometry and related techniques. J ChromatogrA, 2012, 1259:3-15.
[15] Sun W, Tong L, Miao J, et al. Separation and analysis of phenolicacids from Salvia miltiorrhiza and its related preparations by off-line two-dimensional hydrophilic interaction chromatography × reversed- phase liquid chromatography coupled with ion trap time-of-flight mass spectrometry. J Chromatogr A, 2016, 1431:79-88.
[16] Wang L, Liu S, Zhang X, et al. A strategy for identification and structural characterization of compounds from Gardenia jasminoides by integrating macroporous resin column chromatography and liquid chromatography-tandem mass spectrometry combined with ion-mobility spectrometry. J Chromatogr A, 2016, 1452:47-57.
[17] Pamreddy A, Hidalgo M, Havel J, et al. Determination of antibiotics (tetracyclines and sulfonamides) in biosolids by pressurized liquid extraction and liquid chromatography-tandem mass spectrometry.J Chromatogr A, 2013, 1298:68-75.
[18] Wang M, Carver JJ, Phelan VV, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol, 2016, 34(8):828-837.
Identification of two novel sansanmycin analogues fromknockout strain by HPLC-MS/MS
JIANG Zhi-bo, SHI Yuan-yuan, REN Wei-cong, FAN Jia-hui, LEI Xuan, LI Xing-xing, WANG Li-fei, XIE Yun-ying, HONG Bin
Author Affiliations:Key Laboratory of Biotechnology of Antibiotics, the National Health and Family Planning Commission (NHFPC), Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China
To investigate the sansanmycin analogues produced by theknockout strain.
Thefermentation supernatants of the wild type strain ofsp. SS andknockout strain were extracted by the solid phase extraction cartridges, respectively. The 60% (V/V) MeOH/H2O eluents were analyzed by HPLC-MS/MS. The chemical structures for sansanmycins were deduced by comparison of their MS/MS data to the known sansanmycins in the literatures.
Eight sansanmycin analogues including two new structures were discovered from theknockout strain. Compound SS-MX-7 was found to bear two tyrosines (Tyr) substituted at the AA1and AA4, respectively, while SS-MX-8 contained two phenylalanines (Phe) at the two positions.
Two new sansanmycin analogues were first identified from the fermentation extract ofknockout strain, enriching the structural database of uridyl peptide antibiotics (UPAs). The established HPLC-MS/MS method will facilitate the analysis of novel sansanmycin analogues from other recombinant strains.
Uridyl peptide antibiotics;sp. SS; Sansanmycins; HPLC-MS/MS
國家自然科學基金(81621064、81603006、81630089、81703398);中國醫學科學院醫學與健康科技創新工程(2016-I2M-3- 012);“重大新藥創制”國家科技重大專項(2017ZX09101003-006-011、2015ZX09102007-016)
: XIE Yun-ying, Email: xyying102@163.com; HONG Bin, Email: binhong69@hotmail.com
10.3969/j.issn.1673-713X.2018.01.003
解云英,Email:xyying102@163.com;洪斌,Email:binhong69@hotmail.com
2017-11-24
*同為第一作者
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
