伴發潰瘍的I型先天性厚甲癥一例
2018-01-31 06:38:59王燕飛柳君如劉衛兵
中國麻風皮膚病雜志 2018年1期
王燕飛 柳君如 劉衛兵
臨床資料患者,女,38歲。自6歲起四肢伸側及各關節處反復起斑塊、潰瘍、結痂,未診治。15歲時出現指趾甲變厚變硬,手足多汗。自幼聲音嘶啞,未經診治。家族中無類似病史,父母否認近親結婚。體格檢查:一般情況可,智力發育均正常,營養中等,全身淋巴結未觸及腫大。頭無畸形,毛發未見異常。皮膚科查體:雙眼瞼外翻,瞼結膜充血雙眼瞼部分睫毛脫落,瞼球結膜黏連、角膜白斑一側可見胬肉樣增生組織(圖1)。口唇、臀部見大小不等的暗紅色肥厚性瘢痕,中央多個深而大的潰瘍面,部分愈合后形成角化性斑塊(圖2、3);四肢關節伸側以膝關節為重散在角化性斑塊、丘疹,部分呈皮角樣,未見水皰;雙手足指、趾甲板增厚、粗糙、質硬、翹起,呈灰褐色(圖4,5)。掌跖無明顯角化。全部牙齒色黃、磨損變形、左側約第一磨牙處及上顎口腔黏膜可見數片黃豆大白斑,相互融合,心肺功能等未見異常。未見多發性脂囊瘤改變。實驗室及輔助檢查:血、尿、大便常規及生化檢查未見異常,多個指趾甲真菌鏡檢及培養陰性,
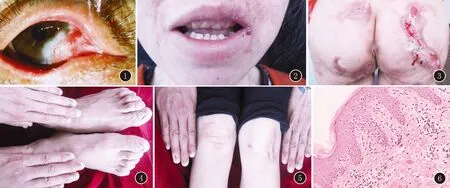
圖1 眼瞼外翻,瞼結膜充血,部分睫毛脫落,瞼球結膜黏連、角膜白斑,左眼玻璃體內見光點圖2 唇部暗紅色瘢痕并潰瘍,牙齒色黃、磨損變形圖3 臀部大小不等的暗紅色肥厚性瘢痕中央多個深而大的潰瘍面,散在角化性斑塊圖4 膝關節散在角化性斑塊、丘疹,部分呈皮角樣,未見水皰圖5 雙手足指、趾甲板增厚、粗糙、質硬、翹起,呈灰褐色圖6 表皮棘層增厚,輕度棘細胞間水腫,真皮血管周圍漿細胞灶狀浸潤(HE,×200)
潰瘍處細菌培養陰性。RPR陰性。組織病理示:表皮棘層增厚,輕度棘細胞間水腫,真皮血管周圍漿細胞呈灶狀浸潤(圖6),抗酸染色陰性。DIF:表皮細胞間及基底膜IgG、C3、IgM、IgA陰性。眼結膜刮片(青島眼科醫院):鏡下可見上皮細胞多量,部分破碎,中性粒細胞多量占40%,淋巴細胞多量占60%。眼超聲(青島眼科醫院):左眼玻璃體內見光點,A超回聲中低,后運動明顯,后界膜高起。
診斷:先天性厚甲癥(I型)
討論先天性厚甲癥(Pachyonychia congenita, PC)為一少見的外胚葉缺陷病,系常染色體顯性遺傳,但也有常染色體隱性遺傳的報道,于1906年由Jadasshon及Lewandowsky首先報道,特點是甲營養不良和不同程度的外胚葉發育不良[1]。男女發病率大致相等,臨床主要表現為指趾甲的過度角化增厚及其他外胚葉缺陷的癥狀。根據臨床表現,可將PC分為三型:I型臨床表現為厚甲、掌跖角化、毛囊角化和口腔黏膜白斑;II型除了I型的臨床特征外,另伴有多發性脂囊瘤、胎生牙和多汗等[2];III型,除了I型和II型的臨床特征外,另伴有角膜白斑和口角干燥等。不同類型的致病基因不同,PC-1是由角蛋白K16或角蛋白K6a基因突變所致,PC-2是由角蛋白K6b或角蛋白K17基因突變所致。本例患者除了指趾甲變厚變硬,手足多汗、口腔黏膜白斑、聲音嘶啞外等經典I型PC特征以外,還有四肢伸側處角化性斑塊,并且臨床上以不易愈合的多發疼痛性潰瘍及瘢痕形成、皮膚黏連作為重要主訴及皮損特征,考慮該例患者基因突變較尋常PC基因突變位點多,既往多篇文獻報道均未提及不易愈合的疼痛性潰瘍及瘢痕形成、皮膚粘連,僅報道有水皰形成[3,4],這是否為I型先天性厚甲癥的補充性特征,其相關突變基因位點等問題有待進一步深入研究。此外,該患者厚甲的發生時間為15歲,符合遲發性厚甲綜合征的臨床特征。值得注意的是,本患者雙眼結膜損害明顯,瞼球黏連進行性加重,影響視物,筆者認為可能與眼瞼、結膜均為外胚層來源而使得炎癥累及該處。
[1] Odom BR, James DW, Berger GT. Andrews disease of the skin clinical dermatology[M]. 9th ed. Philadelphia: Saunders,2000.718-721.
[2] Lucker GPH, van de Kerkh of PCM, Steijlen PM. The hereditary palmoplantar keratoses: an updated review and classification[J]. Br J Dermatol,1994,131(1):11-14.
[3] 黃莉寧,林云冰,薛汝增,等.先天性厚甲癥II型一家系四例[J].國際皮膚性病學雜志,2012,38(3):205-206.
[4] 黃清華,林敏,楊陽,等.先天性厚甲癥1例[J].臨床皮膚科雜志,2012,41(1):49-50.
