QuEChERS-四極桿/靜電場軌道阱高分辨質譜測定動物性食品中氟蟲腈及其代謝物殘留
2017-12-27 02:46:06尹帥星陳達煒方從容趙云峰
分析測試學報 2017年12期
呂 冰,尹帥星,陳達煒*,方從容,周 爽,趙云峰
(1.國家食品安全風險評估中心/衛生部食品安全風險評估重點實驗室,北京 100021;2.島津技邇(上海)商貿有限公司,北京 100027)
QuEChERS-四極桿/靜電場軌道阱高分辨質譜測定動物性食品中氟蟲腈及其代謝物殘留
呂 冰1,尹帥星2,陳達煒1*,方從容1,周 爽1,趙云峰1
(1.國家食品安全風險評估中心/衛生部食品安全風險評估重點實驗室,北京 100021;2.島津技邇(上海)商貿有限公司,北京 100027)
基于低溫處理和QuEChERS方法,采用超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜建立了動物性食品中氟蟲腈及其代謝物殘留的分析檢測方法。樣品采用乙腈提取,經低溫處理,N-丙基乙二胺(PSA)和C18粉分散固相萃取(d-SPE)凈化,以BEH C18色譜柱為分析柱,乙腈-0.1%乙酸溶液為流動相進行梯度洗脫分離,外標法定量。采用高分辨質譜平行反應監測(PRM)掃描模式,以負離子采集進行定性篩查和定量分析。氟蟲腈及其代謝物在0.02~2 μg/L和0.2~20 μg/L質量濃度范圍內均呈良好的線性關系,相關系數(r2)大于0.992。對液體或半液體樣品(如牛奶和雞蛋)和固體樣品(如雞肉),方法的定量下限分別為0.1 μg/kg和 0.2 μg/kg。在不同濃度的加標水平下,氟蟲腈及其代謝物在雞蛋中的平均回收率為81.6%~96.9%,相對標準偏差(RSD)為1.3%~11.5%;在雞肉中的平均回收率為81.2%~96.0%,RSD為3.4%~11.4%;在牛奶中的平均回收率為79.1%~100.1%,RSD為1.5%~10.7%。該方法簡單、靈敏、準確,適用于動物性食品中氟蟲腈及其代謝物的快速篩查和定量檢測,方法的靈敏度滿足歐盟的殘留限量要求。
氟蟲腈;動物性食品;QuEChERS;高分辨質譜;代謝物
氟蟲腈是一種苯基吡唑類殺蟲劑,主要用于殺滅鱗翅目和直翅目的害蟲以及在土壤中的鞘翅目害蟲的幼蟲,同時用以消除蚤、虱、蜱、蟑螂及螨等昆蟲[1]。近期,歐洲多國相繼報道在雞蛋中超標檢出殺蟲劑“氟蟲腈”事件,并導致歐盟多國近千萬枚雞蛋下架[2]。由于短期大量攝入氟蟲腈會對神經系統產生不良影響,而長期攝取氟蟲腈或會導致肝臟、甲狀腺等受損[3-4],歐盟已明令禁止氟蟲腈用于人類食品產業鏈的畜禽養殖過程,并規定氟蟲腈(氟蟲腈和氟蟲腈砜之和,以氟蟲腈計)在雞蛋中的最大殘留限量(MRL)為0.005 mg/kg。國際食品法典委員會(CAC)規定氟蟲腈在肉、蛋、奶中的MRL均為0.02 mg/kg。我國也制定了氟蟲腈在蔬菜、谷類等制品中的MRL,并將氟蟲腈的殘留物定義為氟蟲腈、氟甲腈(MB46513)、氟蟲腈砜(MB46136)、氟蟲腈亞砜(MB45950)之和[5],但暫無在動物性食品中的MRL規定。因此,建立動物性食品中氟蟲腈及其代謝物的高效、快速的殘留檢測方法,以應對可能因“氟蟲腈”事件引發的雞蛋、雞肉和牛奶等動物性食品中氟蟲腈及其代謝物的檢測需要,對保障動物性相關食品安全具有重要意義。
氟蟲腈及其代謝物的測定方法主要有氣相色譜法(GC)[6]、氣相色譜-質譜法(GC-MS)[6-8]、液相色譜法(HPLC)[9]及液相色譜-串聯質譜法(LC-MS/MS)[10-12]。但這些檢測方法均是應用于植物性樣品等基質,而動物性樣品的檢測分析方法鮮見報道。Zhang等[13]利用C18固相萃取柱結合液相色譜-串聯質譜技術建立了雞蛋和雞肉中氟蟲腈的分析方法,但該方法未對氟蟲腈的代謝物進行檢測。世界糧農組織(FAO)在氟蟲腈的評估報告中指出氟蟲腈在動物體內的氟蟲腈母體絕大部分轉化為氟蟲腈砜,后者在蛋黃、皮和脂肪中濃度最高[14]。因此,氟蟲腈母體在動物性樣品中檢出含量較低,而氟蟲腈砜或其它代謝物將是檢測關注的重點。近年來,QTOF及Orbitrap等高分辨質譜檢測技術在農獸藥等化學污染物的檢測分析中得到廣泛應用[15-17]。然而,高分辨質譜技術對目標物的定量檢測分析主要采用全掃描模式(Full scan,FS),與三重四極桿的多反應監測模式(MRM)相比,無法獲得較佳的儀器方法靈敏度。在前期研究中,本課題組應用四極桿/靜電場軌道阱高分辨質譜(Q Exactive)的其它定量掃描模式,如靶向單一離子監測(Target single ion monitoring,TSIM)[18-19]和平行反應監測(Parallel reaction monitoring,PRM)[20],改善了儀器方法的靈敏度,可與三重四極桿的MRM模式相媲美。
本研究采用QuEChERS(Quick、Easy、Cheap、Effective、Rugged、Safe)方法,以乙腈作為提取溶劑,經鹽析后,提取液經低溫冷凍處理和分散固相萃取(d-SPE)凈化,基于超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜的PRM模式,首次建立了動物性食品中氟蟲腈及其代謝物殘留的分析方法。方法的靈敏度滿足歐盟的殘留限量要求,為動物性食品中氟蟲腈及其代謝物的高效、快速測定提供了技術保障。
1 實驗部分
1.1 儀器、試劑與材料
超高效液相色譜-四極桿-靜電場軌道阱高分辨質譜儀(Q-Exactive,美國賽默飛公司)、組織勻漿機(德國博朗公司)、渦旋混合器(美國Scientific Industries公司)、超聲波清洗器(寧波科生儀器廠)和冷凍離心機(德國Sigma公司)。
乙腈為色譜純(美國Fisher Scientific公司),乙酸為HPLC級(美國Tedia公司);氯化鈉和無水硫酸鈉為分析純(國藥試劑有限公司);d-SPE凈化管,內含N-丙基乙二胺(PSA)50 mg、C18粉50 mg和無水硫酸鈉250 mg(島津技邇(上海)商貿有限公司);氟蟲腈、氟甲腈、氟蟲腈砜和氟蟲腈亞砜標準品(純度>98%)購自德國Dr.Ehrenstorfer公司;實驗用水為重蒸水。實際樣品(雞蛋、雞肉、豬肉、豬肝和牛奶等)采購于北京各大超市和農貿市場。
1.2 標準溶液的配制
分別準確稱取氟蟲腈及其代謝物標準品0.01 g(精確至0.000 1 g)于不同的10 mL容量瓶中,用乙腈溶解并定容,配成1 000 mg/L的標準儲備溶液,于-20 ℃儲存。氟蟲腈及其代謝物標準混合中間液(10 mg/L)和標準混合使用液(200 μg/L和20 μg/L)由乙腈逐級稀釋氟蟲腈及其代謝物標準儲備液制得。準確吸取氟蟲腈及其代謝物標準混合使用液(20 μg/L和200 μg/L)0.01、0.02、0.05、0.1、0.2、0.5、1.0 mL于10 mL容量瓶中,用50%乙腈水溶液定容,即得0.02~2 μg/L和0.2~20 μg/L質量濃度范圍的兩套標準工作液,臨用現配。
1.3 試樣制備
取代表性動物性樣品,用組織勻漿機充分攪碎打勻,取其中200 g分裝入潔凈容器中,密封,于-20 ℃保存。
1.4 試樣提取
1.4.1液體與半液體試樣準確稱取樣品5.0 g(精確至0.001 g),置于50 mL離心管中,渦旋混合30 s,加入10 mL乙腈,超聲提取15 min,加入2 g氯化鈉和6 g無水硫酸鈉,渦旋混合30 s,以9 500 r/min于4 ℃離心10 min,轉移上清液于15 mL離心管,待凈化。
1.4.2固體試樣準確稱取樣品2.0 g(精確至0.001 g),置于50 mL離心管中,加水3 mL,渦旋混合30 s,以下操作程序同“1.4.1”,并將上清提取液置于-20 ℃超低溫冰箱中低溫冷凍處理2 h后,待凈化。
1.5 試樣凈化
準確吸取提取液1 mL于2 mL的 d-SPE凈化管中,渦旋混合30 s,取上清液0.5 mL,加水定容至1 mL,過有機微孔濾膜后,待測定。
1.6 儀器分析條件
色譜條件:BEH C18色譜柱(1.7 μm,2.1 mm×100 mm),柱溫:40 ℃;流動相:A為0.1%乙酸溶液,B為乙腈,梯度洗脫,流速0.3 mL/min。梯度洗脫程序:0~6 min,55%~70% B;6~7 min,70%~100% B;7~8 min, 100% B;8~8.5 min,100%~55% B;8.5~11 min,55% B。進樣體積:5 μL。
質譜參數:采用HESI離子化方式;噴霧電壓為3.2 kV;毛細管溫度為320 ℃;加熱溫度為300 ℃;鞘氣為40 arb,輔助氣為10 arb;掃描模式為平行反應監測(PRM),負離子采集模式;分辨率采用70 000 FWHM,碰撞能量(NCE)為20%。氟蟲腈及其代謝物的定性、定量離子對見表1。
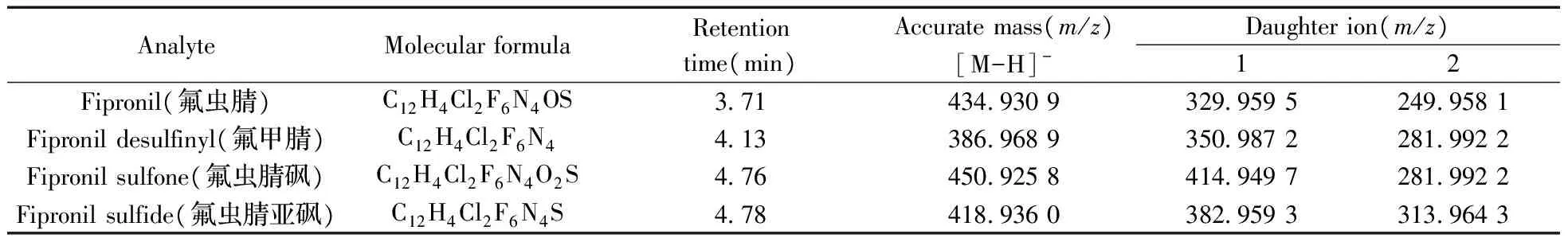
表1 氟蟲腈及其代謝物的母離子和子離子信息Table 1 The information for parent and daughter ions of fipronil and its metabolites
2 結果與討論
2.1 質譜與色譜條件的優化
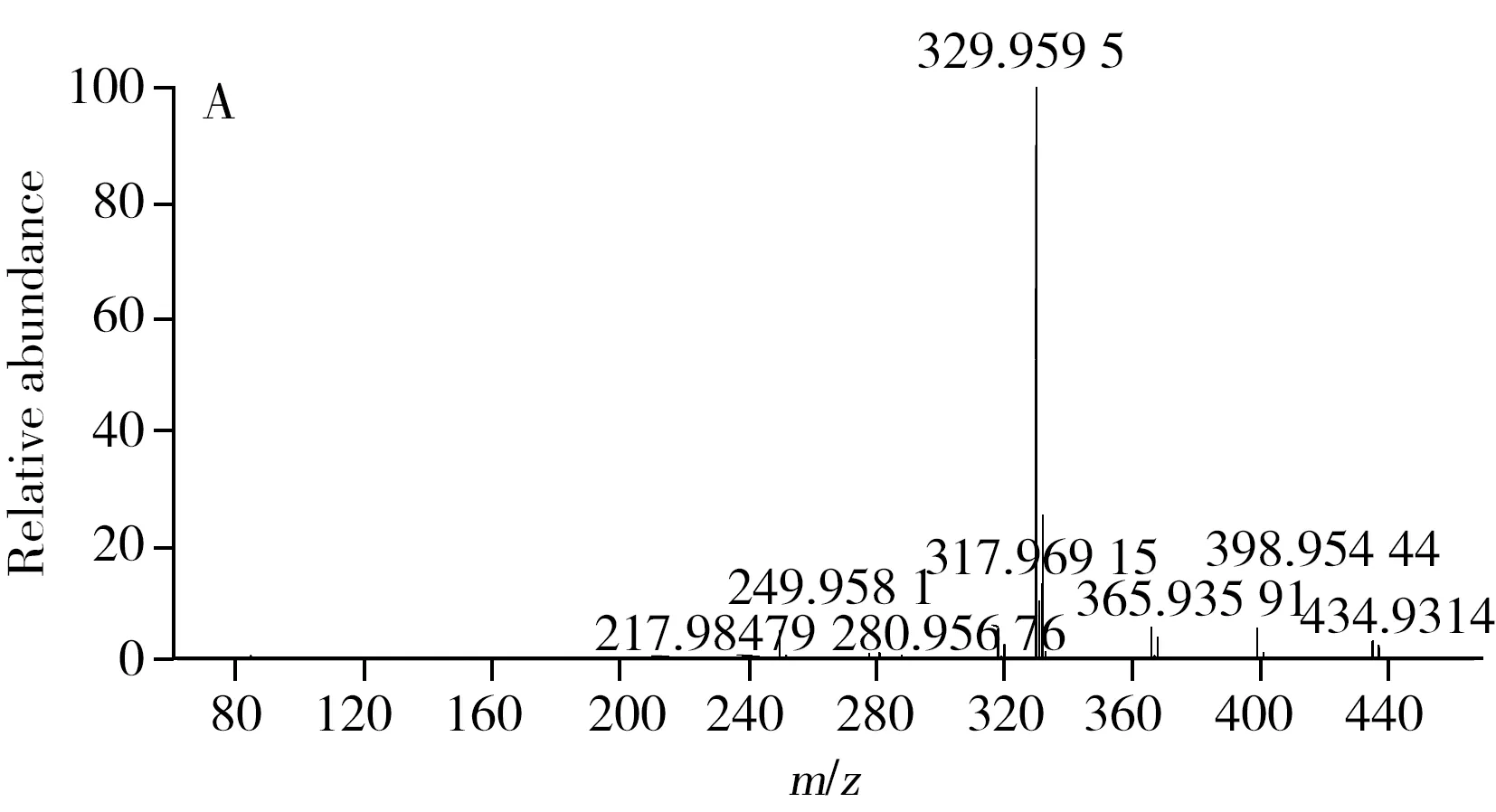
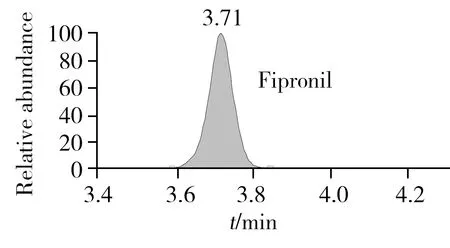
在質譜響應上,氟蟲腈及其代謝物的負離子采集模式優于正離子采集模式。本研究比較了Q Exactive高分辨質譜的FS、TSIM和PRM 3種定量采集模式。其中,FS采集模式是高分辨質譜最為常用的定量模式,該模式是對一定質量范圍內的所有母離子進行全掃描,TSIM采集模式是對單一母離子進行掃描,而PRM采集模式是先從四極桿中選擇目標母離子,然后將其送入高能碰撞池進行二級質譜碎裂,最后以母離子->子離子的精確質量數離子對進行定量,與三重四極桿以離子對為定量手段相似,但其高分辨質譜的檢測能力增強了定性確證的能力,同時由于該模式可通過監測其子離子的精確質量數進一步降低背景噪音響應,從而提高了儀器的靈敏度。因此,本研究采用PRM模式對氟蟲腈及其代謝物進行定性和定量采集。在PRM模式下,本研究進一步對二級質譜的碰撞能量(NCE)進行優化,結果顯示,氟蟲腈及其代謝物在20%的NCE下能夠獲得最佳的二級質譜定量離子響應。圖1A為氟蟲腈母離子在20%的NCE下所采集的二級質譜圖譜,該圖譜包含了未被碎裂的母離子精確質量數(m/z434.931 4)與二級質譜子離子的精確質量數(m/z329.959 5,249.958 1),而圖1B則為該農藥的二級質譜定量子離子(m/z329.959 5)的提取離子色譜圖。
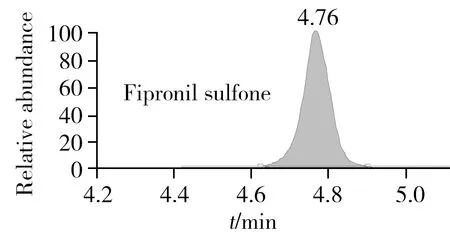
本研究采用BEH C18色譜柱,考察了氟蟲腈及其代謝物在乙腈-水、乙腈-0.1%甲酸或乙酸水溶液、乙腈-5 mmol/L甲酸銨或乙酸銨水溶液為流動相體系下的峰形及質譜響應效果。結果表明,氟蟲腈及其代謝物在中性或偏堿性流動相體系下峰寬更寬,且氟蟲腈砜在酸性流動相體系下具有更好的質譜響應,該現象與文獻報道相符[21];而在酸性流動相體系下,鑒于流動相過低的pH值會抑制氟蟲腈及其代謝物的響應,因此以乙腈和0.1%乙酸水溶液作為流動相體系。圖2為此流動相體系下所獲得的氟蟲腈及其代謝物的色譜圖。
2.2 樣品前處理方法的選擇
目前,乙腈作為提取溶劑具有較高的沉淀蛋白能力,對親脂性目標物具有較好的提取效果,同時有助于下一步QuEChERS的凈化,因而在動物性食品中具有較好的應用[22],對于固體樣品(如雞肉),需預先加入3 mL 水并對其充分混勻,再用乙腈進行提取,以提高對目標物的提取效率。QuEChERS前處理方法由Anastassiades等[23]于2003年開發,其基于d-SPE技術被廣泛應用于農產品中農藥多殘留檢測。本研究采用PSA和C18對樣品提取液進行凈化,其中PSA能有效去除提取液中的極性物質、有機酸和脂肪酸等雜質;而C18對親脂性雜質,尤其是脂肪等物質具有較好的吸附效果。此外,本研究取1 mL樣品提取液進行凈化,50 mg的PSA和50 mg的C18足以達到凈化效果,當進一步增加吸附劑填料對凈化效果并無明顯改善,同時當C18的填料加到100 mg時,對親脂性的氟蟲腈及其代謝物會有一定程度的吸附,導致回收率下降7%~12%。因此,本研究采用內含50 mg的PSA、50 mg的C18和250 mg無水硫酸鈉的d-SPE凈化管進行凈化。
雞肉等固體動物性食品基質較為復雜,脂肪含量較高,單純依靠d-SPE凈化無法滿足去除脂肪的需要。因此為更好地去除脂肪,本研究在進行d-SPE凈化前,先將提取液置-20 ℃超低溫冰箱中低溫冷凍處理,并對冷凍處理時間進行了考察(30 min、1 h、2 h、6 h)。結果發現,提取液冷凍處理2 h后可以達到較好的去除效果,而增加處理時間對去除效果并無明顯改進,故選擇在d-SPE凈化前先對雞肉提取液進行2 h的冷凍處理。
2.3 基質效應
按照“1.4”和“1.5”方法將試樣提取凈化,制備雞蛋、雞肉和牛奶3種空白基質提取液,對空白基質提取液進行分析,確定其不含有痕量目標化合物。用50%的乙腈水溶液及雞蛋、雞肉和牛奶3種空白基質提取液分別配制濃度范圍為0.02~2 μg/L和0.2~20 μg/L的兩套混合標準溶液并繪制標準曲線。本研究依據基質標準溶液和溶劑標準溶液之間的標準曲線斜率比值評價基質效應,斜率值越接近1.0說明基質效應越弱。當斜率比值在0.8~1.2范圍,認為該方法的基質效應可以接受[24]。本方法在低濃度和高濃度標準曲線濃度范圍內對氟蟲腈及其代謝物的基質效應評價如表2所示,雞蛋、雞肉和牛奶的斜率比值范圍在0.9~1.1之間,表明本方法中氟蟲腈及其代謝物在雞蛋、雞肉和牛奶中存在弱的基質效應,基質效應對測定結果的影響可以忽略不計,因而本實驗采用溶劑標準曲線進行定量。
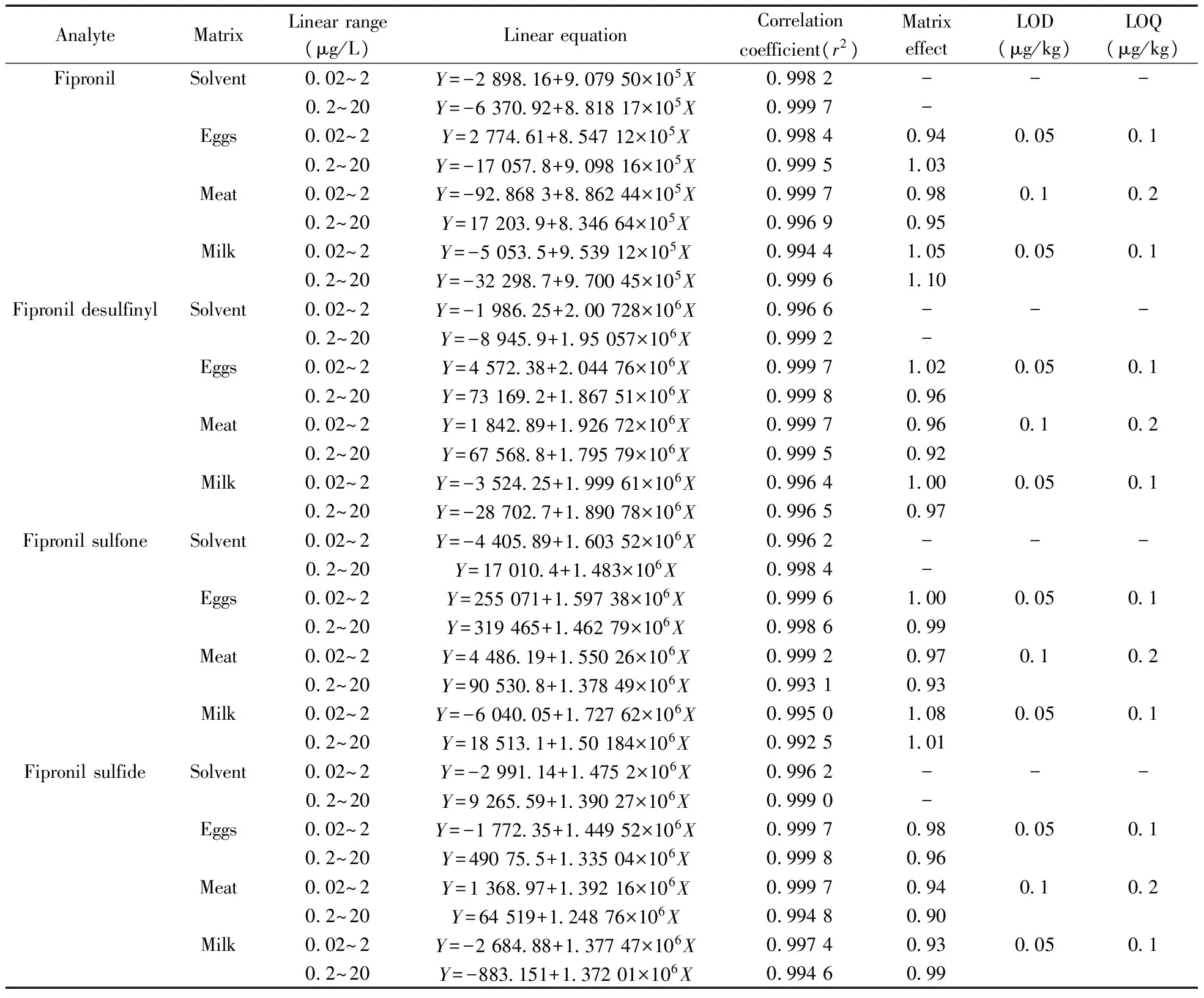
表2 氟蟲腈及其代謝物的線性、基質效應、檢出限及定量下限Table 2 Linear relationship,matrix effect,LOD and LOQ of fipronil and its metabolites
-:no data
2.4 線性范圍與定量下限
本研究配制兩套氟蟲腈及其代謝物的低高濃度系列混合標準溶液,質量濃度范圍分別為0.02~2 μg/L和0.2~20 μg/L,以滿足不同濃度水平下實際樣品和加標樣品的檢測。按“1.6”條件進樣標準溶液,以標準溶液濃度(X,μg/L)為橫坐標,母離子->子離子對的精確質量數的提取離子色譜峰峰面積(Y)為縱坐標,繪制標準曲線,外標法定量。結果表明,氟蟲腈及其代謝物在0.02~2 μg/L和0.2~20 μg/L質量濃度范圍內均呈良好的線性關系,相關系數(r2)>0.992。以空白樣品低水平加標測試方法的檢出限和定量下限,以3倍信噪比(S/N=3)對應的濃度為方法檢出限,以10倍信噪比(S/N=10)對應的濃度為方法的定量下限。表2結果顯示,液體或半液體樣品(如牛奶和雞蛋)取樣量5 g時,氟蟲腈及其代謝物的方法檢出限(LOD)為0.05 μg/kg,定量下限(LOQ)為0.1 μg/kg;固體樣品(如雞肉)取樣量2 g時,氟蟲腈及其代謝物的方法檢出限為0.1 μg/kg,定量下限為0.2 μg/kg。該方法的定量下限均低于歐盟所制定的蛋類中氟蟲腈的最大殘留限量(5 μg/kg)和CAC所制定的蛋類中氟蟲腈的最大殘留限量(20 μg/kg)。
2.5 準確度與精密度
本研究取3種代表性樣品(雞蛋、雞肉和牛奶)進行加標回收實驗,以回收率考察方法的準確度,回收率平行測試結果的相對標準偏差(RSD)考察方法的精密度。加標水平的選擇以接近定量下限水平的0.2 μg/kg為低加標水平,以歐盟制定的蛋類中氟蟲腈的最大殘留限量5 μg/kg為中加標水平,以CAC制定的蛋類中氟蟲腈的最大殘留限量20 μg/kg為高加標水平。取雞蛋、雞肉和牛奶3種空白樣品分別添加低、中和高加標水平的混合標準溶液后,搖勻,放置一定時間后,按照本方法處理,每個濃度水平重復5次,其回收率和RSD結果見表3。由表3可見,氟蟲腈及其代謝物在雞蛋中的平均回收率為81.6%~96.9%,RSD為1.3%~11.5%;在雞肉中的平均回收率為81.2%~96.0%,RSD為3.4%~11.4%;而在牛奶中的平均回收率為79.1%~100.1%,RSD為1.5%~10.7%。
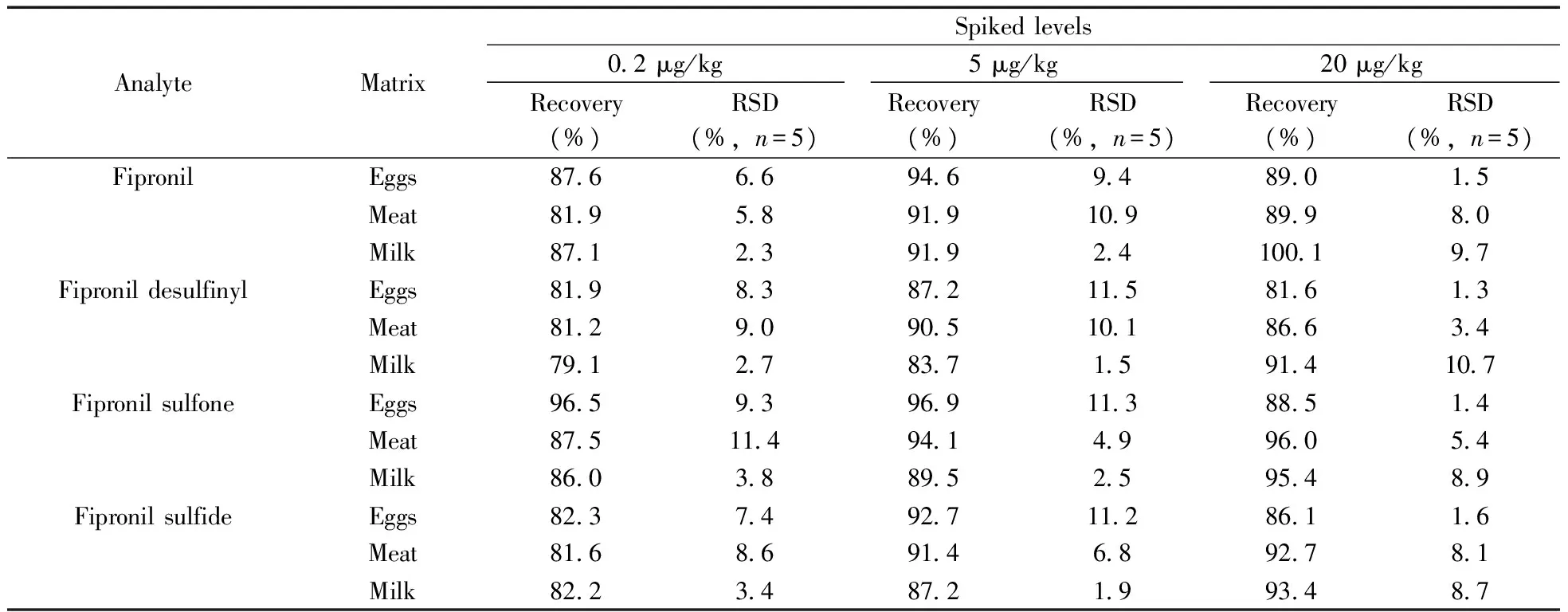
表3 不同加標水平下的回收率和相對標準偏差Table 3 Recovery and relative standard deviation of different spiked levels
2.6 實際樣品的測定
采用本方法檢測市售31份雞蛋、3份雞肉、6份豬肉、4份豬肝和5份牛奶等樣品中的氟蟲腈及其代謝物殘留,結果顯示,在雞肉、豬肉、豬肝和牛奶樣品中均未檢測到氟蟲腈及其代謝物,但有8份雞蛋樣品中檢出氟蟲腈砜,含量為0.13~1.1 μg/kg,低于歐盟所規定的最大殘留限量。
3 結 論
本文采用超高效液相色譜-高分辨質譜技術,結合冷凍處理和分散固相萃取凈化,首次建立了動物性食品中氟蟲腈及其代謝物的分析方法,并應用于實際樣品定性和定量的快速篩查分析。本方法具有樣品前處理操作簡單、價格低和基質效應低等特點,有利于快速監測動物性食品中氟蟲腈及其代謝物的殘留情況;高分辨質譜技術的PRM掃描模式與三重四極桿MRM模式相似,但由于其提供精確的碎片質量數,可進一步降低背景噪音的干擾,提高檢測方法的靈敏度。
[1] Tingle C C D,Rother J A,Dewhurst C F,Lauer S,King W J.Rev.Environ.Contam.Toxicol.,2003,167(1):1-66.
[2] The German Federal Institute for Risk Assessment(BfR).Health Assessment of Individual Measurements of Fipronil Levels Detected in Foods of Animal Origin in Belgium.BfR Opinion No.016/2017 of 30 July 2017.
[3] Hurley P M,Hill R N,Whiting R L.Environ.HealthPersp.,1998,106(1):437-445.
[4] Zhang F F,Hong Y Q,Zhang X.Occup.Health.(張芳芳,洪雅青,張幸.職業與健康),2008,24(20):2211-2213.
[5] GB 2763-2016.National Food Safety Standard—Maximum Residue Limits for Pesticides in Food.National Standards of the People′s Republic of China(食品安全國家標準——食品中農藥最大殘留限量.中華人民共和國國家標準).
[6] Zhou Y,Xu D M,Chen D J,Zhang Z G,Zheng X H,Fang E H.Chin.J.Chromatogr.(周昱,徐敦明,陳達捷,張志剛,鄭向華,方恩華.色譜),2011,29(7):656-661.
[7] Ramasubramanian T,Paramasivam M,Jayanthi R,Chandrasekaran S.FoodChem.,2014,150(1):408-413.
[8] Yan S Y,Li X H,Zhao E C,Jia C H.Chin.J.Anal.Lab.(閆思月,李興海,趙爾成,賈春虹.分析試驗室),2016,35(4):386-389.
[9] Bai B Q,Li M P,Zhang S W.FoodSci.(白寶清,李美萍,張生萬.食品科學),2014,35(24):254-268.
[10] Lin T,Fan J L,Yang D S,Liu X Y,Shao J L,Li Y G,Liu H C.J.Instrum.Anal.(林濤,樊建麟,楊東順,劉興勇,邵金良,李彥剛,劉宏程.分析測試學報),2015,34(12):1360-1365.
[11] Du Y Y,Luo Y L,Wang J Q,Wu D M,Zhai Y Z,Yin X Y,Xu N S.Environ.Chem.(堵燕鈺,羅漪漣,王潔瓊,吳冬梅,翟云忠,殷雪琰,徐牛生.環境化學),2017,36(4):928-930.
[12] Liu Z Z,Qi P P,Wang X Y,Wang Z W,Xu X H,Chen W X,Wu L Y,Zhang H,Wang Q,Wang X Q.FoodChem.,2017,230(1):423-431.
[13] Zhang M Y,Bian K,Zhou T,Song X Q,Liu Q Y,Meng C Y,He L M.J.Chromatogr.B,2016,1014(1):31-36.
[14] FAO.Pesticide Residues in Food.Fipronil(202).2001:191-365.
[15] Sun B X,Guo D H,Ding Z P,Ji F,Dong J C,Yao J T.J.Instrum.Anal.(孫碧霞,郭德華,丁卓平,冀峰,董吉川,姚勁挺.分析測試學報),2010,29(10):1017-1024.
[16] Kaserzon S L,Heffernan A L,Thompson K,Mueller J F,Ramos M J G.Chemosphere,2017,182(1):656-664.
[17] Chen D W,Lü B,Zou J H,Yang X,Miao H,Zhao Y F.J.Instrum.Anal.(陳達煒,呂冰,鄒建宏,楊欣,苗虹,趙云峰.分析測試學報),2014,33(12):1327-1333.
[18] Chen D W,Zhao Y F,Miao H,Wu Y N.J.Chromatogr.A,2014,1374(1):268-272.
[19] Chen D W,Zhao Y F,Miao H,Wu Y N.Talanta,2015,134(1):144-152.
[20] Chen D W,Miao H,Zou J H,Cao P,Ma N,Zhao Y F,Wu Y N.J.Agric.FoodChem.,2015,63(2):485-492.
[21] He M,Song D,Dong F S,Zheng Y Q.Environ.Chem.(賀敏,宋丹,董豐收,鄭永權.環境化學),2016,35(5):925-932.
[22] Gong X M,Hua M M,Wang H T,Wang B J,Ma R H.J.Instrum.Anal.(宮小明,華萌萌,王洪濤,王炳軍,馬榮檜.分析測試學報),2017,36(7):897-901.
[23] Anastassiades M,Lehotay S J,Stajnbaher D,Schenck F J.J.AOACInt.,2003,86:412-431.
[24] Besil N,Cesio V,Heinzen H,Fernandez-Alba A R.J.Agric.FoodChem.,2017,65(23):4819-4829.
Determination of Fipronil and Its Metabolites in Animal Derived Foods by QuEChERSMethod Combined with Ultra Performance Liquid Chromatography-High-resolution Benchtop Q Exactive Mass Spectrometry
Lü Bing1,YIN Shuai-xing2,CHEN Da-wei1*,FANG Cong-rong1,ZHOU Shuang1,ZHAO Yun-feng1
(1.China National Center for Food Safety Risk Assessment/Key Laboratory of Food Safety Risk Assessment,Ministry of Health,Beijing 100021,China;2.Shimadzu-GL Sciences(Shanghai)Laboratory Supplies Co.,Ltd.,Beijing 100027,China)
Based on cryogenic treatment and QuEChERS method,an ultra performance liquid chromatography-high-resolution benchtop Q Exactive mass spectrometry method for the determination of fipronil and its metabolites in animal derived foods was established.The samples were extracted with acetonitrile,and cleaned up by the cryogenic treatment and dispersive solid phase extraction(d-SPE)with the N-propylethlene diamine(PSA)and C18.The chromatographic analysis was performed on a BEH C18column with 0.1% acetic acid and acetonitrile as mobile phase by gradient elution,and the external standard calibration was used for quantification.The negative ion acquisition mode was applied,and the quantitative analysis was carried out by high resolution mass spectrometry using parallel reaction monitoring(PRM)mode.Fipronil and its metabolites showed good linearities in the concentration ranges of 0.02-2 μg/L and 0.2-20 μg/L,with the correlation coefficients(r2)above 0.992.For liquid or semi-liquid samples(such as milk and eggs)and the solid samples(such as chicken),the limits of quantification(LOQs)for 4 analytes were between 0.1 μg/kg and 0.2 μg/kg.At different spiked levels,the average recoveries of fipronil and its metabolites in eggs,chicken and milk were in the ranges of 81.6%-96.9%,81.2%-96.0% and 79.1%-100.1%,with the relative standard deviations(RSDs)of 1.3%-11.5%,3.4%-11.4% and 1.5%-10.7%,respectively.The method was simple,sensitive and accurate,and was suitable for the rapid screening and quantitative analysis of fipronil and its metabolites in animal derived foods,and the sensitivity of the method could meet the requirement for residual limitation of EU.
fipronil;animal derived food;QuEChERS;high-resolution mass spectrometry;metabolites
2017-08-22;
2017-08-30
國家食品安全風險評估中心高層次人才隊伍建設523項目
*
陳達煒,博士,副研究員,研究方向:食品衛生,Tel:010-67779768,E-mail:chendw@cfsa.net.cn
10.3969/j.issn.1004-4957.2017.12.002
O657.7
A
1004-4957(2017)12-1424-07
