斷節參抑制α-葡萄糖苷酶活性成分研究
2017-12-09 07:59:01毛坤軍張莉周慧云黃平
中國現代中藥 2017年11期
毛坤軍,張莉,周慧云,黃平
(江西醫學高等專科學校,江西 上饒 334000)
斷節參抑制α-葡萄糖苷酶活性成分研究
毛坤軍,張莉,周慧云,黃平*
(江西醫學高等專科學校,江西 上饒 334000)
目的尋找斷節參中具有抑制α-葡萄糖苷酶活性的成分。方法采用體外抑制α-葡萄糖苷酶活性模型進行追蹤,用各種色譜法分離,根據理化性質和譜學數據鑒定結構,篩選出活性較強的單體化合物并進行酶活性抑制動力學研究。結果斷節參乙醇提取物的乙酸乙酯溶性部位具有顯著的抑制α-葡萄糖苷酶活性,從中分離出3個化合物,其中斷節參苷H和青陽參苷B兩個皂苷類化合物具有較強抑制α-葡萄糖苷酶活性,IC50分別為21.90、35.32 mg·L-1,明顯高于陽性對照藥阿卡波糖(IC50=1 017.41 mg·L-1)。酶活性抑制動力學反應結果表明,兩個皂苷對α-葡萄糖苷酶的抑制類型均為非競爭性抑制劑。結論斷節參苷H和青陽參苷B為首次報道對α-葡萄糖苷酶有抑制活性。
斷節參;α-葡萄糖苷酶;斷節參苷H;青陽參苷B;抑制類型
斷節參系蘿藦科鵝絨藤屬植物昆明杯冠藤CynanchumwallichiiWight的根,又名對節參,青洋參,斷節參主要分布于我國四川、貴州、云南以及廣西等地。具有補肝腎、強筋骨之功效,主要用于治療風濕關節炎及腎虛腰痛、病后體虛、跌打損傷[1]等。現代化學研究表明,斷節參主要含有C21甾體皂苷、苯乙酮、甾醇等類化合物[2-3],現代藥理活性研究發現其具有抗炎鎮痛、抗氧化、抗腫瘤、抗抑郁等藥理活性[4-6],但未見對α-葡萄糖苷酶抑制作用的研究報道。
α-葡萄糖苷酶抑制劑可競爭性抑制小腸內α-葡萄糖苷酶的活性,延緩或抑制葡萄糖在腸道的吸收,從而有效降低餐后高血糖。由于其獨特的優勢,目前已被廣泛用于糖尿病及其并發癥的防治。為進一步開發利用斷節參資源,本實驗采用體外抑制α-葡萄糖苷酶活性篩選模型,從斷節參乙醇提取物的乙酸乙酯活性部位分離得到12個化合物,其中兩個皂苷類化合物具有明顯的抑制α-葡萄糖苷酶活性。
1 主要儀器和試劑
680型自動酶標儀(美國BIO-RAD公司);AY220電子分析天平(日本島津分析儀器公司);雷磁PHSJ-3F型pH計(上海精密科學儀器有限公司),Costar 96孔細胞培養板。
對硝基苯酚(p-Nitrophenol,33920)購于上海阿拉丁有限公司;α-葡萄糖苷酶(α-glucosidase,SLBB7613V)、4-硝基苯-α-D-吡喃葡萄糖苷(4-ntrophenyl-α-D-glucopyranoside,PNPG,BCBG2931V)、阿卡波糖(acarbose,Lot16869)、DMSO均購自Sigma公司;乙醇、石油醚、二氯甲烷、乙酸乙酯、正丁醇、甲醇均為分析純(廣東華光科技股份有限公司)。
斷節參藥材采自云南昆明(批號:120914),經南京中醫藥大學陳建偉教授鑒定為蘿藦科鵝絨藤屬植物昆明杯冠藤CynanchumwallichiiWight的根。
2 方法
2.1 提取與分離
斷節參藥材20 kg,打成粗粉,以8倍量95%乙醇回流提取2 h,重復2次,再以50%乙醇回流提取提取1次,合并提取液濾液,濃縮至無醇味,再加水混懸依次用石油醚、乙酸乙酯、正丁醇萃取,得石油醚部位230 g、乙酸乙酯部位420 g、正丁醇部位480 g,剩余水部位950 g。
體外α-葡萄糖苷酶抑制活性研究發現,石油醚和乙酸乙酯溶性部位有較強活性。取乙酸乙酯部位經硅膠(200~300目)柱色譜(依次以石油醚-乙酸乙酯和二氯甲烷-甲醇系統梯度洗脫)分離得到不同極性段,再經過中壓制備柱色譜、ODS 柱色譜(甲醇-水系統梯度洗脫)、Sephadex LH-20柱色譜(甲醇-水)反復分離和純化。最終分離得到化合物1(128 mg)、2(79 mg)和3(37 mg)。
2.2 抑制α-葡萄糖苷酶活性成分的篩選方法
2.2.1 檢測方法 本檢測在96孔板上進行,反應體系參照康文藝等[7]的方法,按照公式(1)計算抑制率(I)。
I(%)=[1-(A樣品-A樣品空白)/(A陰性-A空白]×100%
(1)
并用Origin軟件求出相應半數最大抑制濃度(IC50)值。同時設定陰性對照組(緩沖液+酶液+底物)、空白對照組(緩沖液)、樣品測定組(樣品+酶液+底物)、樣品對照組(樣品+緩沖液)。
2.2.2 標準曲線制作 根據反應體系,用磷酸緩沖液(pH 6.8)配制1000 μmol·L-1的PNP,稀釋成 400、300、200、150、100、50、25、5、0 μmol·L-1。分別取7種不同濃度的PNP溶液各160 μL,加入0.2 moL·L-1Na2CO3溶液80 μL,混勻,在405 nm下測定A值,平行3次。以A值為縱坐標,對硝基苯酚濃度為橫坐標,做出標準曲線。
2.2.3α-葡萄糖苷酶活力的測定 根據反應體系:[110 μL 磷酸鉀緩沖液(pH 6.8),加入20 μL 0.2 U·mL-1α-葡萄糖苷酶,10 μL DMSO,37 ℃恒溫15 min后加入2.5 mmoL·L-1PNPG 20 μL,37 ℃恒溫反應15 min,再加入80 μL 0.2 moL·L-1Na2CO3溶液],于405 nm波長下測定A值。
酶活力單位定義:37 ℃、pH 6.8條件下,每分鐘水解底物所產生1 μmoL對硝基苯酚的酶量,規定為1個酶活力單位(U)。
3 結果與討論
3.1 結構鑒定
化合物1:白色無定型粉末(甲醇),Liebermann-Burchard和Keller-Kiliani反應陽性,UV光譜在220nm有最大吸收,推測其可能為甾體2-去氧糖苷類化合物。1H-NMR(DMSO-d6,300 MHz)δ:1.04(3H,s,19-CH3),1.53(3H,s,18-CH3),2.03(3H,s,21-CH3),3.72(3H,s,3″′-OCH3),3.79(3H,s,3″″-OCH3),4.05(1H,brs,3α-H),4.55(1H,dd,J=12,4.5 Hz,12α-H),4.70(1H,d,J=9.3 Hz,1″″-H),4.74(1H,d,J=9.3 Hz,1″′-H),4.86(1H,d,J=8.7 Hz,1″-H),5.28(1H,brs,6-CH),5.56(1H,s,2′-CH),6.81(2H,d,J=8.7 Hz,4′,6′-H),7.72(2H,d,J=8.7 Hz,3′,7′-H),10.26(1H,s,5′-OH);13C-NMR(DMSO-d6,300 MHz)δ:38.2(C-1),28.8(C-2),76.3(C-3),38.0(C-4),137.9(C-5),118.7(C-6),33.7(C-7),73.0(C-8),42.9(C-9),36.2(C-10),23.8(C-11),72.1(C-12),57.0(C-13),88.4(C-14),33.0(C-15),32.9(C-16),91.1(C-17),10.1(C-18),17.5(C-19),209.1(C-20),27.2(C-21),163.8(C-1′),120.4(C-2′),131.2(C-3′,7′),115.1(C-4′,6′),161.7(C-5′)。此外,100.8(C-1″)、36.1(C-2″)、79.8(C-3″)、74.9(C-4″)、71.5(C-5″)、18.0(C-6″)、56.4(OCH3);98.8(C-1″′)、35.2(C-2″′)、76.4(C-3″′)、81.7(C-4″′)、68.0(C-5″′)、17.9(C-6″′)、57.8(OCH3);95.0(C-1″″)、37.9(C-2″″)、66.1(C-3″″)、81.9(C-4″″)、67.4(C-5″″)、18.0(C-6″″)分別為β-D-Ole、β-D-Cym、β-D-Dgt碳信號。以上數據與文獻[4]報道的斷節參苷H基本一致,故化合物1鑒定為斷節參苷H。
化合物2:白色無定型粉末(甲醇),Liebermann-Burchard和Keller-Kiliani反應陽性,UV光譜在220nm有最大吸收,推測其可能為甾體2-去氧糖苷類化合物。1H-NMR(DMSO-d6,300 MHz)δ:0.99,1.02(6H,d,J=6.9 Hz,5′,6′-CH3),1.04(3H,s,19-CH3),1.40(3H,s,18-CH3),2.03(3H,s,7′-CH3),2.09(3H,s,21-CH3),3.31(3H,s,3″-OCH3),3.33(3H,s,3′″-OCH3),3.34(3H,s,3″″-OCH3),3.69(1H,m,3α-H),4.33(1H,dd,J=11.7,4.5 Hz,12α-H),4.52(1H,d,J=9.3 Hz,1″″-H),4.67(1H,d,J=9.0 Hz,1″′-H),4.76(1H,d,J=8.7 Hz,1″-H),5.27(1H,brs,6-CH),5.43(1H,s,2′-CH);13C-NMR(DMSO-d6,300 MHz)δ:38.6(C-1),28.7(C-2),75.0(C-3),37.0(C-4),137.9(C-5),118.7(C-6),33.7(C-7),73.0(C-8),43.0(C-9),36.2(C-10),23.7(C-11),71.4(C-12),56.6(C-13),88.3(C-14),33.0(C-15),31.6(C-16),90.9(C-17),9.9(C-18),17.5(C-19),208.7(C-20),26.8(C-21),164.6(C-1′),113.0(C-2′),164.5(C-3′),36.2(C-4′),20.5(C-5′)20.7(C-6′)15.9(C-7′)。此外,100.8(C-1″)、35.5(C-2″)、79.8(C-3″)、75.0(C-4″)、71.5(C-5″)、17.9(C-6″)、56.4(OCH3);99.3(C-1″′)、35.5(C-2″′)、76.6(C-3″′)、81.8(C-4″′)、67.8(C-5″′)、17.9(C-6″′)、56.4(OCH3);95.0(C-1″″)、37.9(C-2″″)、76.5(C-3″″)、82.0(C-4″″)、67.9(C-5″″)、18.0(C-6″″)、57.8(OCH3)分別為β-D-Ole、β-D-Cym、β-D-Cym碳信號。以上數據與文獻[4]報道的青陽參苷B基本一致,故化合物2鑒定為青陽參苷B。
化合物3:白色針狀結晶,mp 215~220 ℃,1H-NMR(C5D5N,300 MHz)δ:1.46(3H,s,19-CH3),2.00(3H,s,18-CH3),2.64(3H,s,21-CH3),3.88~3.99(2H,m,3α和12α-H),5.38(1H,brs,6-H);13C-NMR(C5D5N,300 MHz)δ:39.3(C-1),32.1(C-2),71.6(C-3),43.5(C-4),140.4(C-5),118.8(C-6),34.3(C-7),74.4(C-8),45.0(C-9),37.4(C-10),29.5(C-11),69.0(C-12),60.5(C-13),89.3(C-14),35.1(C-15),32.8(C-16),92.6(C-17),9.4(C-18),18.6(C-19),209.6(C-20),27.9(C-21)。以上數據與文獻[2]報道的去酰基蘿藦苷元基本一致,故鑒定化合物3為去酰基蘿藦苷元。
3.2 提取部位對α-葡萄糖苷酶活性抑制成分的篩選
斷節參提取物不同極性部位及陽性藥阿卡波糖的α-葡萄糖苷酶活性抑制作用見圖1,石油醚、乙酸乙酯、總皂苷部位的抑制活性較好,其抑制率明顯高于其他部位以及陽性對照藥,所以可采用活性追蹤方式對乙酸乙酯部位繼續分離。

注:各提取物初篩質量濃度均為1.5 g·L-1。圖1 不同提取物對α-葡萄糖苷酶抑制活性的比較
3.3 對α-葡萄糖苷酶活性抑制作用化合物的篩選
對分離得到的化合物進行初步α-葡萄糖苷酶活性抑制篩選,結果見表1。皂苷類化合物具有明顯的α-葡萄糖苷酶活性抑制作用,而苷元對α-葡萄糖苷酶活性基本沒有抑制作用。

表1 單體化合物體外抑制α-葡萄糖苷酶活性
3.4 活性化合物IC50實驗
不同質量濃度(200、100、50、25、12.5、6.25 μg·mL-1)下斷節參苷H和青陽參苷B抑制α-葡萄糖苷酶活性見圖2。根據圖2并用Origin軟件得斷節參苷H和青陽參苷B的IC50分別為21.90、35.32 mg·L-1,遠小于陽性對照藥阿卡波糖的IC50值(IC50=1 017.41 mg·L-1),且兩個化合物對α-葡萄糖苷酶活性抑制作用均與濃度呈正量效關系。
3.5 受試化合物抑制類型的確定
化合物1和2分別取適宜的兩個不同質量濃度,PNPG取4個不同質量濃度,分別測定反應速度。按Lineweave-Burk作圖法,以1/[S](L·mmol-1)為橫坐標,1/V為縱坐標,分別繪制兩個化合物的抑制作用動力學曲線(見圖3和4)。圖3和4顯示化合物1和2對α-葡萄糖苷酶活性抑制作用均屬于非競爭性抑制劑,反應速度Vmax隨受試化合物濃度增大而減小,米氏常數Km值保持不變。
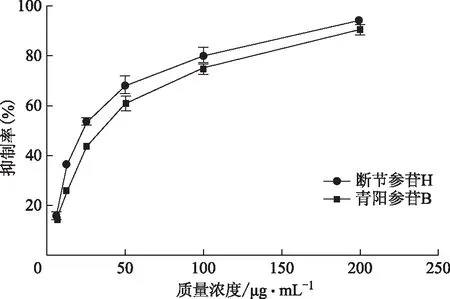
圖2 受試化合物不同質量濃度下對α-葡萄糖苷酶抑制曲線

圖3 化合物1的Lineweave-Burk雙倒數曲線

圖4 化合物2的Lineweave-Burk雙倒數曲線
4 討論
近年來,鑒于皂苷類化合物毒副作用小、作用溫和、性質穩定的優點[7],其降血糖作用研究進展迅速,如苦瓜皂苷、大豆皂苷、人參皂苷等三萜皂苷以及藠頭皂苷、玉竹皂苷、三七皂苷等甾體皂苷均具有顯著的降血糖作用[8-10]。C21甾體皂苷是斷節參中主要化學成分,其是否有降血糖功效筆者未見相關文獻報道,本實驗首次利用體外抑制α-糖苷酶活性篩選模型對斷節參提取物抑制α-葡萄糖苷酶活性進行了初步篩選,結果表明乙酸乙酯部分有較高的抑制活性,采用活性追蹤方法從斷節參中分離得到12個以皂苷和二苯酮類為主的活性化合物,并對其進行體外抑制α-葡萄糖苷酶活性篩選,結果表明皂苷類化合物具有顯著活性,而皂苷苷元及二苯酮基本沒有活性。
為了驗證實驗結果,本實驗還通過D-101大孔樹脂富集得到總皂苷部位,結果發現在同等質量濃度下其抑制α-葡萄糖苷酶活性又明顯高于乙酸乙酯部位,進一步證明了斷節參降血糖主要活性成分是皂苷類成分。本課題組將繼續對乙酸乙酯部位進行分離,以期得到更多皂苷類化合物,為皂苷類化合物降血糖作用機制、構效關系以及體內降血糖效果的研究奠定基礎。
[1] 云南省食品藥品監督管理局.云南省中藥材標準[S].昆明:云南科學技術出版社,2005:489.
[2] 張壯鑫,周俊.斷節參苷的結構[J].化學學報,1983,41(11):1058.
[3] 張壯鑫,周俊.昆明杯冠藤的化學成分[J].云南植物研究所,1982,4(4):413.
[4] 陳剛,裴月湖.昆明杯冠藤和Pfaffia glomerate的化學成分研究[D].沈陽:沈陽藥科大學,2009:16.
[5] 康文藝,張麗,宋艷麗.滇丁香中抑制α-葡萄糖苷酶活性成分研究[J].中國中藥雜志,2009,34(4):406.
[6] 張壯鑫,周俊.昆明杯冠藤的化學成分[J].云南植物研究所,1982,4(4):413.
[7] 張晟,陳祥貴.降血糖植物皂苷研究進展[J].中藥材,2007,30(5):616.
[8] 劉劍青,肖小年.藠頭皂苷的分離純化及其活性研究[D].南昌:南昌大學,2013:50.
[9] 王謙,張璐,邊曉麗,等.α-葡萄糖苷酶抑制劑及構效關系的研究進展[J].中國新藥雜志,2014,23(2):189.
[10] 季芳,肖國春,董莉,等.藥用植物來源的α-葡萄糖苷酶抑制劑研究進展[J].中國中藥雜志,2010,35(12):1633.
StudyonActiveComponentInhibitingα-GlucosidaseInhibitofromCynanchumwallichii
MAOKunjun,ZHANGLi,ZHOUHuiyun,HUANGPing*
(Departmentofpharmacy,JiangxiMedicalCollege,Shangrao334000,China)
Objective:To findα-glucosidase inhibitors fromCynanchumwallichii.MethodsThe α-glucosidase inhibitor was isolated and purified by the bioactivity-guided methodinvitro,which was used to analyze the inhibitory activity against α-glucosidase.The inhibitory kinetic of the isolations was also investigated.ResultsThe ethyl acetate extract ofCynanchumwallichiishowed strong inhibitory activity and three compounds were isolated and identified,only wallicoside H(IC50=21.90 mg·L-1)and otophylloside B showed strong inhibitory activity(IC50=35.32 mg·L-1)which was higher than that of acarbose(IC50=1 017.41 mg·L-1).Both of them showed noncompetitive type on α-glucosidase.ConclusionIt was reported that wallicoside H and otophylloside B have α-glucosidase Inhibitors active for the first time.
Cynanchumwallichii;α-glucosidase;wallicoside H;otophylloside B;inhibition type
*
黃平,講師,研究方向:藥效物質基礎研究;E-mail:737259639@qq.com
10.13313/j.issn.1673-4890.2017.11.009
2017-03-06)
