磺酸樹脂催化甲縮醛一步羰化制高附加值甲氧基乙酸甲酯
2017-10-16 08:12:57石磊姚杰朱文良劉中民
化工學報 2017年10期
關鍵詞:催化劑
石磊,姚杰,朱文良,劉中民
(1中國科學院大連化學物理研究所,遼寧 大連 116023;2沈陽化工大學能源與化工產業技術研究院,遼寧 沈陽 110142)
磺酸樹脂催化甲縮醛一步羰化制高附加值甲氧基乙酸甲酯
石磊1,2,姚杰2,朱文良1,劉中民1
(1中國科學院大連化學物理研究所,遼寧 大連 116023;2沈陽化工大學能源與化工產業技術研究院,遼寧 沈陽 110142)
甲氧基乙酸甲酯(MMAc)是重要的精細化學品,同時是擬開辟的由合成氣間接法制乙二醇的中間產物。鑒于文獻報道的甲縮醛(DMM)氣相羰化制 MMAc存在 CO與 DMM比例過高(>100),CO一次轉化率過低(<0.5%)等缺點,采用釜式反應器,系統研究液相DMM羰基化反應過程中諸多因素,如溶劑種類、不同牌號磺酸樹脂催化劑、反應溫度、壓力、反應時間、催化劑前處理等因素對DMM轉化率以及產物MMAc選擇性的影響。環丁砜顯著提高了CO在液相中的溶解度,并有效抑制醛基游離以及DMM歧化反應,使DMM最大限度地向羰化反應方向進行。采用H-Y分子篩為催化劑,在反應溫度120℃,初始反應壓力3.0 MPa時,MMAc選擇性僅為5%,而以磺酸樹脂為催化劑時,MMAc選擇性可以達到45%,說明磺酸樹脂催化劑比H-Y分子篩具有更強的羰化能力。DMM在H-Y分子篩微孔表面更易發生歧化反應,導致二甲醚選擇性大于90%。DMM轉化率隨反應溫度、壓力的增高和反應時間的增長而增大,MMAc選擇性隨反應時間增長和壓力增高而提高、隨著反應溫度的升高先提高后降低。隨著催化劑前處理溫度升高,樹脂催化劑中吸附的水含量逐漸減少,MMAc選擇性逐步提高,但過高溫度會導致催化劑孔道塌陷、表面結焦,羰化效果變差。以環丁砜為溶劑,D-009B樹脂為催化劑,反應溫度110℃、壓力5 MPa、反應6 h,DMM轉化率接近100%,MMAc選擇性達到74.32%,顯示較好的工業應用前景。
甲縮醛;甲氧基乙酸甲酯;羰基化反應;磺酸樹脂;環丁砜;催化
Abstract:Methyl methoxyacetate (MMAc),an important fine chemical,is an intermediate product producing ethylene glycol from syngas.Literature reports showed that high molar ratio of CO to DMM (>100) and low CO conversions (<0.5%) were obviously beneficial for vapor-phase carbonylation of dimethoxymethane (DMM) to MMAc.Using a slurry phase reactor,this work systematically studied the effects of different solvents,sulfonic acid resin catalysts,reaction temperature,pressure,reaction time and drying temperature on DMM conversion and MMAc selectivity.The solubility of CO in liquid phase was significantly increased by sulfolane solvent ,thus obviously promoting the carbonylation reaction and suppressing the hydration as well as disproportionationreactions of DMM.Comparing with H-Y,the carbonylation activity of sulfonic acid resin catalyst was higher.The high rate of DMM disproportionation for H-Y was attributed to the micropores of zeolite,which facilitated a critical initial step in the formation of DME and MF.With the increased reaction temperature,pressure or reaction time,the conversion of DMM increases,whereas the selectivity of MMAc first increased and then decreased.With increasing the drying temperature,the moisture content left in the resin gradually decreased.But higher drying temperature resulted in the collapse of pores and surface sintering.For the liquid phase carbonylation process,many complicated and unreported side reactions were accompanied.The DMM conversion can reach to about 100% with 74.32% MMAc selectivity using sulfolane as solvent and D-009B as the catalyst at 110℃ and 5 MPa for 6 h.It was anticipated that this direct carbonylation of DMM to produce MMAc process was promising for industrial manufacture.
Key words:dimethoxymethane; methyl methoxyacetate; carbonylation; sulfonic acid resion; sulfolane; catalysis
引 言
甲氧基乙酸甲酯(MMAc)是重要的精細化工原料,可用于手性胺類化合物的動力學拆分、維生素B6及磺胺-5-嘧啶等合成、聚合反應催化劑等[1],市場價格高,為 35000~40000元/噸。同時,MMAc是擬開辟由甲縮醛(DMM)制備乙二醇的重要中間產物,由 MMAc經過加氫反應可得到乙二醇單甲醚,得到的乙二醇單甲醚和水發生取代反應(甲氧基由羥基取代)即可制得大宗化工原料乙二醇。目前乙二醇制備途徑是石腦油裂解得到乙烯,乙烯經過部分氧化得到環氧乙烷,環氧乙烷經水合反應最終得到乙二醇[2-5]。但是,隨著石油資源日益枯竭,采用煤或者天然氣為原料制成合成氣(CO+H2),再通過間接法合成乙二醇的非石油路線逐漸引起人們廣泛關注[6-10],其中從合成氣經草酸二甲酯加氫后制得乙二醇路線近年來報道較多[11-12]。
已經報道的MMAc合成方法有:① 氯乙酸或氯乙酸甲酯和甲醇鈉反應[13],該方法反應原料氯乙酸是由乙酸和氯氣發生取代反應得到,反應過程復雜,且腐蝕嚴重并污染環境,不適合大規模生產;② 乙二醇單甲醚氧化法[14],該方法乙二醇單甲醚來源受限,且氧化工藝復雜,產物為甲氧基乙酸,還需經過額外酯化反應后才能得到 MMAc;③ 甲縮醛(DMM)或甲醛和甲酸或甲酸甲酯偶聯法[15-17],該方法缺點為副產物較多,MMAc選擇性難以突破30%;④ DMM羰基化法。Bell等[7]報道采用雜多酸為催化劑,DMM 為反應原料,發生一步羰基化反應,但產物 MMAc選擇性低于 30%。之后,他們[18]實現了DMM發生氣相羰基化反應,采用的是拓撲結構為 FAU分子篩為催化劑,在低壓(0.3 MPa)、100℃時,在保證高選擇性(79%)前提下,MMAc收率可以達到20%。與DMM羰化的競爭反應是自身歧化反應,生成甲酸甲酯(MF)和二甲醚(DME)[19-21]。Liu等[22]采用不具有微孔結構的固體酸 H-Nafion樹脂為催化劑,通過固定床氣相羰化DMM,成功將 MMAc選擇性提升至 90%。但H-Nafion樹脂售價約100美元/克,價格昂貴,使催化成本增加。
DMM 氣相羰化的優點是工藝簡單,無污染,產物MMAc選擇性較高,一般可以達到70%以上。原料DMM工業來源方便,價格低廉,一般在濃硫酸催化作用下,由甲醇和甲醛在合成塔中反應得到。但DMM氣相羰基化反應也存在如下問題:已報道[18-22]的反應原料DMM以0℃時飽和蒸氣的狀態被CO氣體攜帶進反應裝置,導致DMM進樣量過少;CO與DMM比例過高,一般高于100:1;CO一次轉化率過低,一般低于0.5%。
基于以上原因,采用釜式反應器(經過計算原料 CO 與 DMM 比例為 3/1~5/1)系統研究液相DMM 羰基化反應過程中諸多因素,如溶劑種類、不同磺酸樹脂催化劑、反應溫度、壓力、反應時間以及催化劑前處理等因素對 DMM 轉化率和產物MMAc選擇性的影響。
1 實驗部分
1.1 實驗藥品
甲縮醛(Alfa Aesar,產地上海,≥98%),環丁砜(國藥,產地上海,化學純,水含量≤0.5%),正己烷(富宇試劑,產地天津,≥97%,含有少量水、芳香烴及不揮發物),二氯甲烷(大茂試劑,產地天津,≥99.5%,含有少量水及蒸發殘渣),1,4-二氧六環(國藥,產地上海,≥99.5%,含有少量水及蒸發殘渣),甲苯(產地沈陽,試劑純),丙酮(產地沈陽,試劑純),乙酸甲酯(國藥,產地上海,≥98%),H-Y分子篩(硅鋁比 6.1,產地日本),磺酸樹脂催化劑(D-009B:含水量 50.00%~58.00%,全交換容量≥3.50 mmol·g?1;DA-330:含水量 50.00%~55.00%,全交換容量≥1.00 mmol·g?1,丹東明珠有限公司),高純CO(產地大連,99.999%)。
1.2 實驗流程
稱取3 ml甲縮醛(DMM),30 ml溶劑[正己烷(n-hexane)、甲苯(methylbenzene)、二氯甲烷(dichloromethane)、1,4-二氧六環(1.4-dioxane)或環丁砜(sulfone)]以及 5 g處理后磺酸樹脂催化劑裝入120 ml含有聚四氟內膽的反應釜中。在室溫條件下將純CO通入反應釜,至所需壓力(0.8、1.4、2.0、2.5、3.0、3.5、4.0或5.0 MPa)后開始升溫至所需反應溫度(90、100、110、130或150℃),升溫時間控制在30 min左右,待釜內溫度達到反應溫度后,穩定反應2、4或者6 h。停止反應后約30 min降至室溫,開釜并在反應液中加入0.2 g內標物乙酸甲酯,搖勻后用0.2 μm過濾篩將催化劑過濾,濾液通過氣相色譜進行分析。
1.3 分析測試儀器
反應氣相及液相產物由島津GC-2014C氣相色譜儀定性和定量分析。氣相色譜分析條件如下:HP-FFAP色譜柱,30 m×0.530 mm×1.00 μm;FID氫火焰檢測器,進樣口溫度230℃,檢測器溫度250℃,初始柱溫40℃,并以10℃·min?1速率升至100℃,再以20℃·min?1升溫到220℃,保持10 min。載氣:氮氣,流速155.5 ml·min?1。DMM轉化率 =所有產物折算回DMM質量/起始加入DMM質量;MMAc質量選擇性=生成 MMAc質量/所有產物總質量。
2 實驗結果與討論
2.1 無溶劑分子篩催化DMM羰化反應
采用5 g硅鋁比為6.1的H-Y分子篩為催化劑,反應原料DMM體積50 ml,反應溫度120℃,CO初始壓力3.0 MPa,達到反應溫度120℃時釜內壓力6.1 MPa,反應時間3 h,產物二甲醚(DME)選擇性達到90%以上,甲氧基乙酸甲酯(MMAc)選擇性僅為5%。該結果說明,在無溶劑條件下,CO在純DMM中溶解度過低,導致DMM在分子篩B酸活性位羰化效率較低,在分子篩微孔表面主要發生歧化反應,導致大量DME生成。
2.2 溶劑種類對DMM羰化反應的影響
表1為以5 g磺酸樹脂D-009B為催化劑,3 ml DMM為反應原料,溶劑分別為30 ml甲苯、1,4-二氧六環、正己烷、二氯甲烷或環丁砜,室溫通入3.0 MPa CO,反應溫度110℃,反應時間6 h時,DMM轉化率和各種產物選擇性的結果。

表1 不同反應溶劑對DMM羰化反應結果的影響Table 1 Effect of different solvents on DMM carbonylation reaction
從表1中可看出,相同反應條件,不同溶劑種類,DMM 轉化率和各種產物選擇性差別很大。甲苯為溶劑時,DMM 羰化效果很差,DMM 轉化率僅為13.87%,MMAc選擇性僅為2.16%。DMM主要發生自身歧化反應,生成了大量的甲酸甲酯(MF,25.13%)和 DME(57.33%),見式(2)。生成的 MF部分分解生成CO和甲醇(MeOH),見式(3)。同時,2分子DMM和1分子H2O作用,生成1分子二甲氧基甲醚(DMM2)和2分子MeOH,見式(4),所以產物中有大量 MeOH(10.80%)生成。可見以甲苯為溶劑時,DMM 羰化能力極差,主要發生自身歧化反應,反應主產物為DME。1,4-二氧六環為溶劑時,DMM 轉化率為 51.09%,MMAc質量選擇性為15.99%,產物 DMM2選擇性相對較高為 31.75%,產物乙醇酸甲酯(MG)為16.92%。說明DMM 在溶劑1,4-二氧六環中,在H2O存在條件下,易于發生醛基的游離和插入反應,游離出的甲醛與 H2O和CO發生羰化反應,生成乙醇酸(HGA),HGA又和MeOH發生酯化反應,生成MG,見式(5)和式(6)。正己烷為溶劑時,DMM 轉化率為 80.12%,產物MMAc 質量選擇性為 44.08%,MG為 8.11%,MMAc2為 34.09%,羰化產物(MMAc、MMAc2以及MG均為羰化產物)質量選擇性總和為86.28%。可見,正己烷溶劑利于羰化反應的進行,同時產物中幾乎沒有 DMM2生成也是很好的證明(DMM2基本全部發生羰化反應,生成 MMAc2)。二氯甲烷為溶劑時,DMM轉化率為87.43%,產物MMAc 質量選擇性為 46.70%,MG為 15.52%,MMAc2為17.98%。可見,二氯甲烷溶劑也利于羰化反應的進行。與正己烷溶劑相比,二氯甲烷溶劑更易于促進DMM 游離出甲醛,因此產物中 MG選擇性高約7.41%,但MMAc2選擇性低約16%。環丁砜為溶劑時,DMM幾乎全部轉化(98.70%),并且MMAc選擇性能夠達到71.91%,MG選擇性為11.11%,產物中其他副產物均較少 (低于18%),產物中未檢測到DMM2和MMAc2生成。可見,環丁砜極易促進DMM發生直接羰化反應,CO溶解能力較強。相比其他溶劑,環丁砜能有效抑制醛基游離以及插入反應,保證反應原料DMM能最大限度發生羰化反應,因此環丁砜是 DMM 發生羰基化反應生成 MMAc的優良溶劑。

2.3 不同類別固體酸對DMM羰化反應影響
分別采用5 g硅鋁比為6.1的H-Y分子篩和5 g磺酸樹脂D-009B為催化劑,溶劑環丁砜,反應溫度110℃,室溫下通入3 MPa CO,反應溫度達到110℃時釜內壓力分別為3.8 MPa和3.6 MPa,反應時間 6 h。以 H-Y為催化劑時,DMM 轉化率僅為39.52%,產物分別為:63.38% DME,28.12% MeOH,6.29% DMM2以及2.21% MMAc。與2.1節所述一致,說明無論有無溶劑,液相DMM主要在分子篩微孔表面發生歧化反應,生成大量 DME。而以D-009B為催化劑時,DMM 轉化率為 98.70%,MMAc選擇性可達 71.91%。以上結果充分說明,磺酸樹脂類催化劑與分子篩相比,更適合DMM發生液相羰基化反應。
2.4 反應溫度對DMM羰化反應影響
圖1(a)為以DA-330為催化劑,室溫下通入2.5 MPa CO,反應溫度分別為 90、100、110、130或150℃,達到反應溫度時釜內壓力分別為3.2、3.2、3.2、3.4和3.6 MPa時DMM轉化率和MMAc選擇性隨時間變化結果。隨著反應溫度逐步升高,DMM轉化率逐漸增加,當反應溫度高于110℃時,DMM轉化率均高于 90%,當反應溫度高于 130℃時,DMM接近完全轉化。當反應溫度為90℃時,產物MMAc的選擇性僅為 28.00%,隨著反應溫度逐漸升高(90~110℃),MMAc選擇性逐漸增加,110℃時,MMAc選擇性達到最大(47.26%),繼續提高反應溫度(110~150℃),MMAc的選擇性逐漸下降,150℃時,MMAc選擇性為40.42%。
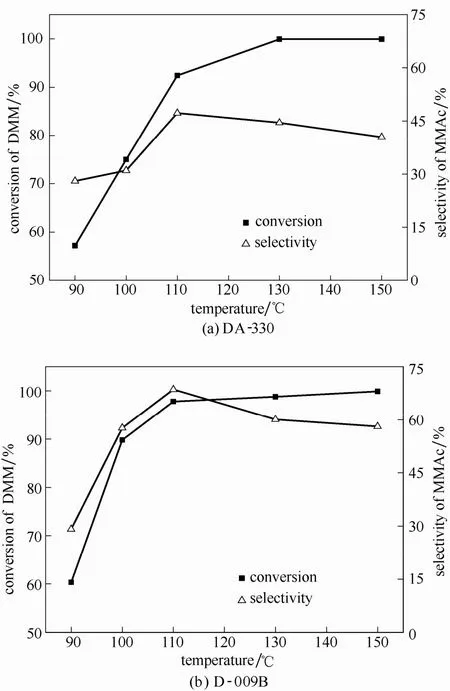
圖1 反應溫度對不同樹脂催化劑DMM羰化反應的影響Fig.1 Effect of reaction temperature on DMM carbonylation for DA-330 and D-009B catalysts
當以D-009B為催化劑時,反應結果如圖1(b)所示,DMM 的轉化率與各產物的選擇性隨反應溫度變化趨勢與 DA-330催化劑基本一致。但采用D-009B時,在相同反應溫度,產物MMAc選擇性比DA-330高約20%,說明D-009B與DA-330相比,在相同溫度下有更強的定向羰化能力。
推測采用樹脂類催化劑,在低溫和高溫段MMAc選擇性較低的原因是:低溫時,催化劑羰化活性較低,反應原料在少量 H2O(反應溶劑、原料DMM以及樹脂催化劑中攜帶)存在前提下,更容易發生醛基游離和插入反應,生成DMM2和MeOH。高溫時,游離出的甲醛(HCHO)在溶劑中移動速度更快,增加分子間相互接觸概率,兩個甲醛分子能夠發生二聚反應,生成MF。歧化生成的MF在高溫時又發生分解反應,生成MeOH和CO。隨著反應溫度升高,DMM 游離出甲醛能力增強,而高溫又容易導致游離出的甲醛與H2O和CO發生羰基化反應生成HGA,之后又與MeOH發生酯化反應,生成MG。因此,MG選擇性隨反應溫度的升高而增加,更多游離醛基被羰化消耗,也是導致MMAc選擇性降低的原因之一。
另外,由于采用樹脂類催化劑,當反應溫度過高時催化劑存在失活現象,過高的溫度會使催化劑內部C—S鍵斷裂,磺酸基團脫落。同時,過高反應溫度還容易使催化劑表面產生結焦,生成的大分子或高分子產物聚積于催化劑表面酸中心,阻礙反應物接近。過高溫度還可能造成聚苯乙烯磺酸樹脂催化劑內部孔道發生塌陷,導致反應物無法接觸到活性中心,從而極大影響催化活性。因此,在該反應條件下,最佳反應溫度為110℃。
2.5 反應壓力對DMM羰化反應影響
以DA-330為催化劑時,當室溫下分別通入壓力為 0.8、1.4、2.0、2.5、3.0、3.5、4.0或 5.0 MPa CO,反應溫度110℃時釜內反應壓力分別為1.2、1.8、2.8、3.2、3.8、4.2、4.8和6.0 MPa,圖2(a)為DMM轉化率和MMAc選擇性隨壓力變化結果。當通入壓力為0.8 MPa時,DMM轉化率為78.40%,MMAc選擇性僅為 9.07%。隨著反應壓力升高(0.8~3.0 MPa),DMM 轉化率逐漸增加(78.40%~94.10%),MMAc選擇性顯著提高(9.07%~57.59%),升高約48.52%。繼續增加反應壓力(3.0~5.0 MPa),DMM轉化率增加緩慢(94.10%~99.90%),同時MMAc選擇性也提升緩慢(57.59%~61.00%)。
當以D-009B為催化劑時,反應結果如圖2(b)所示,DMM轉化率和MMAc選擇性變化趨勢與以DA-330基本一致,即隨著CO壓力逐漸升高,DMM轉化率和 MMAc選擇性都隨之增加。但使用D-009B時,在相同反應壓力條件下,產物MMAc選擇性比DA-330高約15%以上,說明D-009B具有更好的DMM羰基化能力。在CO壓力僅為0.8 MPa時,MMAc選擇性仍高于50%,說明即使較少量的 CO溶解在環丁砜中,D-009B仍能夠促使DMM發生高效羰化反應。

圖2 反應壓力對不同樹脂催化劑DMM羰基化反應的影響Fig.2 Effect of reaction pressures on DMM carbonylation for DA-330 and D-009B catalysts
DMM 轉化率隨反應壓力增大而增加的原因是:隨著反應壓力逐步提升,CO在溶劑環丁砜中的溶解度逐漸增加,即有更多的CO溶解在液相環丁砜中,就使得催化劑的酸中心同時接觸DMM和CO的概率增加,所以反應原料DMM轉化率隨反應壓力的增加而逐步提高。MMAc選擇性隨反應壓力增大而增加的原因是:低壓時,由于溶解在環丁砜中的CO量較少,在催化劑活性位接觸的CO與DMM的比例較低,導致DMM羰基化效率較低,更多發生歧化反應。隨著CO與酸中心接觸概率增大(即 CO 壓力升高),羰化效率顯著提高,MMAc選擇性提升迅速。但初始反應壓力高于3.5 MPa時,MMAc選擇性增加緩慢,原因是高壓時更有利于促進游離的甲醛發生羰基化反應,導致MG選擇性增加較快。部分醛基被消耗是導致MMAc在高壓時選擇性增加緩慢的主要原因。
2.6 反應時間對DMM羰化反應影響
圖3(a)為以DA-330為催化劑,在室溫下通入3.5 MPa CO,當反應溫度達到110℃時釜內壓力為4.2 MPa,反應時間分別為1、2、4、6或8 h時,DMM轉化率和MMAc、MeOH以及MG選擇性隨反應時間變化的結果。隨著反應時間增加(1~8 h),DMM轉化率逐漸增加,當反應時間超過4 h時,DMM轉化率均高于90%,當反應時間8 h時,DMM接近完全轉化(99.9%)。1 h 時,MMAc選擇性僅為 28.20%,隨著反應時間增加(1~4 h),MMAc選擇性快速增長,4 h時,MMAc選擇性為50.29%,持續增加反應時間(4~8 h),MMAc選擇性增長相對緩慢,8 h時,MMAc選擇性達到最大值(58.56%)。
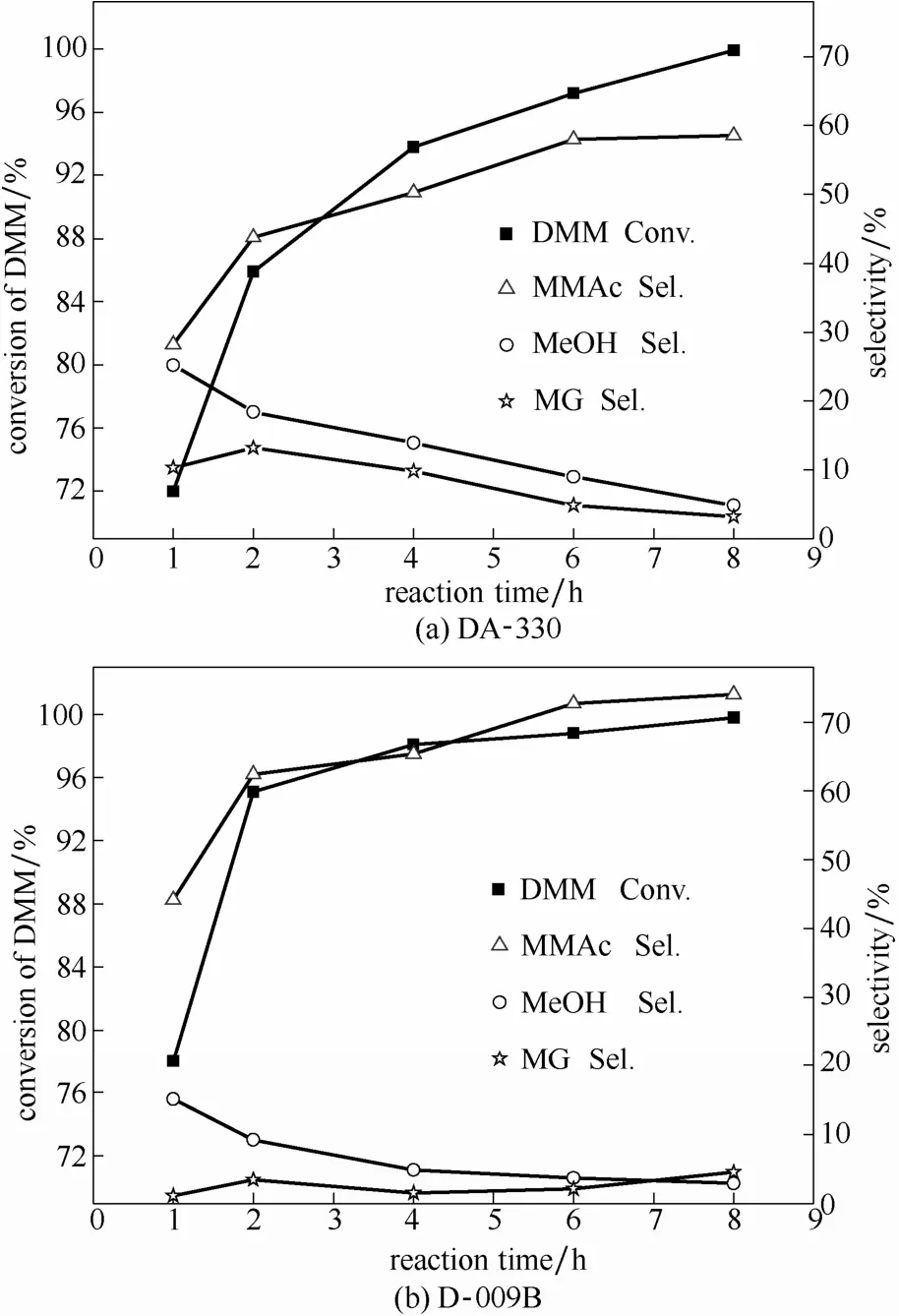
圖3 反應時間對不同樹脂催化劑DMM羰基化反應的影響Fig.3 Effect of reaction time on DMM carbonylation for DA-330 and D-009B catalysts
造成 MMAc選擇性在不同反應時間段變化的原因是:反應時間較短時,DMM轉化率較低,DMM更容易和溶劑中少量 H2O發生可逆反應,因此MeOH選擇性較高(25.20%)。隨著反應時間增加(從1 h至8 h),發生反應的DMM量逐漸增加,DMM羰化反應不是可逆反應,因此剩余DMM的量逐漸減少,式(4)向逆方向進行,產物中MeOH選擇性隨反應時間增長而減少(由25.20%降至4.80%),參與直接羰化的DMM逐漸增加,所以MMAc選擇性隨反應時間的增加而升高。
當以D-009B為催化劑時,反應結果如圖3(b)所示,隨反應時間變化,D-009B對DMM羰化反應影響與DA-330影響規律基本一致。但采用D-009B時,在相同反應時間MMAc的選擇性比DA-330高約15%,短時間反應(1~4 h),以D-009B為催化劑時產物中 MeOH的選擇性要低于 DA-330(低約10%),MG 的選擇性要低于 DA-330(低約 10%)。MeOH與MG的生成均需要H2O的參與,由此推測DA-330相較于 D-009B催化劑能夠吸附更多的H2O(包括物理吸附和化學吸附),導致 H2O參與的副反應更容易發生,因此直接羰化能力相對較弱。
2.7 催化劑前處理對DMM羰化反應影響
圖4(a)為以DA-330為催化劑,在室溫下通入3.5 MPa CO,反應溫度達110℃時釜內壓力為 4.2 MPa,催化劑前處理條件分別為 90、110、120或130℃烘箱中鼓風干燥 6 h時,DMM 轉化率及MMAc選擇性反應結果。隨著前處理溫度升高(90~120℃),DMM 轉化率逐漸增加,當前處理溫度為120℃時,DMM轉化率接近100%。繼續升高前處理溫度至 150℃,DMM 轉化率反而略有下降,MMAc選擇性的變化趨勢和DMM轉化率變化規律基本一致。
以不同前處理溫度(90~130℃)處理的 DA-330為催化劑,各產物選擇性如表2所示,表2中經干燥后樹脂催化劑剩余水含量計算公式為
resin water content=(0.5mresin?Δm) /mresin
式中,0.5mresin表示生產廠家給出的樹脂水含量約為樹脂總質量的50%(質量),Δm表示經過烘箱不同溫度干燥后樹脂減少質量(通過稱重法測定)。
當催化劑前處理溫度較低時,樹脂催化劑內仍然含有大量的水(包含物理吸附的水與化學吸附的結晶水),認為影響產物選擇性高低的關鍵就是體系中水含量的多少。首先,反應過程中水可以和磺酸樹脂B酸位上H+相互作用(氫鍵),顯著降低催化劑活性,因此催化劑前處理溫度較低時(90℃),樹脂中水含量10.14%(質量),DMM轉化率僅為74.0%。其次,水含量越多越容易與DMM反應,生成DMM2和MeOH。DMM羰化反應和DMM與H2O反應是競爭反應,H2O含量越高越不利于羰基化反應進行,所以催化劑前處理溫度越低(90℃),產物MMAc選擇性越低(9.4%),產物 DMM2(14.2%)和MeOH(28.0%)選擇性相對較高。同時,H2O含量的多少還顯著影響產物MG的生成,因為隨著體系中H2O含量增加能夠促進DMM游離出更多的甲醛,而甲醛與H2O和CO發生羰基化反應生成HGA,HGA又與MeOH反應生成MG和H2O,因此催化劑前處理溫度越低,產物MG選擇性相對越高。而當催化劑前處理溫度過高時(高于130℃),雖然絕大部分吸附的水能夠從樹脂中脫附出去(樹脂中水含量僅0.36%),但過高的前處理溫度容易使樹脂催化劑表面燒結失活,導致羰化能力下降,因此采用高溫處理的樹脂催化劑,MMAc選擇性顯著下降。

表2 DA-330催化劑不同前處理溫度對DMM羰化反應結果的影響Table 2 Influence of different drying temperatures on DMM carbonylation

圖4 不同催化劑前處理溫度對DMM羰化反應的影響Fig.4 Effect of pretreatment temperature on DMM carbonylation for DA-330 and D-009B catalysts
如圖4(b)所示,隨著催化劑前處理溫度變化,D-009B對DMM羰化反應結果的影響與DA-330影響規律基本相同。使用D-009B催化劑時,前處理溫度高于 100℃時,MMAc選擇性均高于 DA-330催化劑(高15%~20%),120℃為兩種磺酸樹脂最佳前處理溫度。
3 結 論
本文系統研究了液相DMM羰基化反應過程中諸多因素如溶劑種類、不同磺酸樹脂催化劑、反應溫度、壓力、反應時間、催化劑前處理等因素對DMM轉化率以及產物MMAc選擇性的影響,結論如下。
(1)溶劑環丁砜能顯著增加CO在液相體系中的濃度,并能有效抑制醛基游離,保證反應原料DMM最大限度地發生羰化反應。
(2)在液相反應中,樹脂催化劑比分子篩具有更強的羰化能力,DMM 在分子篩微孔表面更易發生歧化反應。
(3)DMM轉化率隨著反應溫度、壓力、時間增加而增大;MMAc選擇性隨著反應溫度的增加呈先增加后減小趨勢,隨反應時間和壓力增加而增大。
(4)隨著催化劑前處理溫度升高,樹脂催化劑中吸附的水含量逐漸減少,但高溫度會導致催化劑孔道塌陷、表面結焦等。因此,DMM 轉化率和MMAc選擇性都呈現先增加后減小的趨勢。
(5)液相DMM羰化反應過程中會發生多個復雜副反應,如DMM發生自身歧化反應生成DME和 MF,MF發生分解反應生成 MeOH和 CO,DMM與H2O生成DMM2和MeOH,MeOH發生脫水反應生成 DME,液相 DMM游離出甲醛,甲醛和 DMM2分別發生羰化反應生成 HGA和MMAc2,HGA又和 MeOH發生酯化反應生成MG和H2O等。
[1]BALKENHOHL F,DITRICH K,HAUER B,et al.Optically active aminesvialipase-catalyzed methoxyacetylation[J].Journal Fur Praktische Chemie-practical Applications and Applied Chemistry,1997,339(4): 381-384.
[2]BERTY J M.Ethylene oxide synthesis[J].Applied Industrial Catalysis,1983,(1): 207-236.
[3]YUE H R,ZHAO Y J,GONG J L,et al.Ethylene glycol: properties,synthesis,and applications[J].Chemical Society Reviews,2012,41(11): 4218-4244.
[4]李玉芳,伍小明.國內外乙二醇生產技術進展[J].中國石油和化工,2004,(12): 17-19.LI Y F,WU X M.Progress in ethylene glycol production technology at home and abroad[J].China Petroleum and Chemical Industries,2004,(12): 17-19.
[5]LI B,BAI S Y,WANG X F,et al.Hydration of epoxides on[CoⅢ(salen)]encapsulated in silica-based nanoreactors[J].Angewandte Chemie International Edition,2012,51(46): 11517-11521.
[6]SUN Y,WANG H,SHEN J H,et al.Highly effective synthesis of methyl glycolate with heteropolyacids as catalysts[J].Catalysis Communications,2009,10(5): 678-681.
[7]CELIKF E,LAWRENCE H,BELL A T.Synthesis of precursors to ethylene glycol from formaldehyde and methyl formate catalyzed by heteropoly acids[J].Journal of Molecular Catalysis A: Chemical,2008,288(1): 87-96.
[8]KIM Y G,LEE J S,LEE K H.Catalytic carbonylation for the synthesis of chemical intermediates[J].Research on Chemical Intermediates,1998,24(2): 197-211.
[9]LARSON A T.Process for the preparation of glycolic acid:US2153064[P].1939-04-04.
[10]BARRI S A I,CHADWICK D.Carbonylation of formaldehyde with zeolite catalysts[J].Catalysis Letters,2011,141(6): 749-753.
[11]周建飛,劉曉勤,劉定華.草酸酯法由合成氣制備乙二醇技術研究進展[J].化工進展,2009,28(1): 47-50.ZHOU J F,LIU X Q,LIU D H.Progress and perspective of synthesis of ethylene glycol from syngasviaoxalate[J].Chemical Industry and Engineering Progress,2009,28(1): 47-50.
[12]ZHAO Y J,LI S M,WANG Y,et al.Efficient tuning of surface copper species of Cu/SiO2catalyst for hydrogenation of dimethyl oxalate to ethylene glycol[J].Chemical Engineering Journal,http://dx.doi.org/10.1016/j.cej.2016.12.027.
[13]徐志珍,潘鶴林.甲氧基乙酸甲酯合成工藝研究[J].上海化工,2002,(7): 14-15.XU Z Z,PAN H L.A study on synthetic process of methyl methoxyacetate[J].Shanghai Chemical Industry,2002,(7): 14-15.
[14]聶俊琦,李雄,王亦鳴,等.一種甲氧基乙酸的制備方法:104892390[P].2015-09-09.NIE J Q,LI X,WANG Y M,et al.Method for preparation of methoxyacetic acid: 104892390[P].2015-09-09.
[15]沈鑫權,劉洪忠,高志賢,等.甲縮醛與甲酸偶聯合成甲氧基乙酸甲酯[J].天然氣化工,2012,(6): 37-39.SHEN X Q,LIU H Z,GAO Z X,et al.Synthesis of methy methoxy acetate from methylal and formic acid[J].Natural Gas Chemical Industry,2012,(6): 37-39.
[16]HE D H,HUANG W G,LIU J Y,et al.The activity of H4SiW12O40for the coupling of formaldehyde and methyl formate to methyl glycolate and methyl methoxy acetate[J].Journal of Molecular Catalysis A: Chemical,1999,145(1/2): 335-338.
[17]HUANG W G,HE D H,LIU J Y,et al.Catalytic condensation of formaldehyde and methyl formate over 12-tungstosilicic compounds[J].Applied Catalysis A: General,2000,199(1): 93-98.
[18]CELIK F E,KIM T J,BELL A T.Vapor-phase carbonylation of dimethoxymethane over H-Faujasite[J].Angewandte Chemie International Edition,2009,48(26): 4813-4815.
[19]CELIK F E,KIM T,BELL A T,et al.An investigation into the mechanism and kinetics of dimethoxymethane carbonylation over FAU and MFI zeolites[J].Journal of Catalysis,2010,274(2):150-162.
[20]SHAPOVALOV V,BELL A T.Theoretical study of zeolite-catalyzed dimethoxymethane carbonylation to methyl methoxyacetat[J].Journal of Physical Chemistry C,2010,114(41): 17753-17760.
[21]CELIK F E,KIM T J,BELL A T.Effect of zeolite framework type and Si/Al ratio on dimethoxymethane carbonylation[J].Journal of Catalysis,2010,270(1): 185-195.
[22]LIU S P,ZHU W L,SHI L,et al.A highly efficient Nafion-H catalyst for vapour phase carbonylation of dimethoxymethane[J].Royal Society of Chemistry Advances,2014,4(77): 40999-41002.
Efficient sulfonic acid resin catalysts for carbonylation of dimethoxymethane to value-added methyl methoxyacetate
SHI Lei1,2,YAO Jie2,ZHU Wenliang1,LIU Zhongmin1
(1Institute of Chemical Physics,Chinese Academy of Sciences,Dalian116023,Liaoning,China;2Institute of Industrial Chemistry and Energy Technology,Shenyang University of Chemical Technology,Shenyang110142,Liaoning,China)
O 643.32
A
0438—1157(2017)10—3739—08
10.11949/j.issn.0438-1157.20170327
2017-03-30收到初稿,2017-06-09收到修改稿。
聯系人:石磊,劉中民。
石磊(1982—),男,副教授。
國家自然科學基金項目(21303106);遼寧省自然科學基金項目(2015020243);一等博后基金項目(2014M560224);第八批特別資助博后基金項目(2015T80275)。
Received date:2017-03-30.
Corresponding author:SHI Lei,shilei1982@dicp.ac.cn; Prof.LIU Zhongmin,liuzm@dicp.ac.cn
Foundation item:supported by the National Natural Science Foundation of China (21303106),the Natural Science Foundation of Liaoning Province(2015020243),the First Class General Financial Grant from the China Postdoctoral Science Foundation (2014M560224) and the Special Financial Grant from the China Postdoctoral Science Foundation (2015T80275).
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
