六氮雜[3.3.3]螺槳烷的多位點(diǎn)烷基化反應(yīng)
2017-09-03 05:32:04張俊林王伯周畢福強(qiáng)王錫杰張家榮
火炸藥學(xué)報(bào) 2017年4期
張俊林, 王伯周,2, 畢福強(qiáng), 王錫杰,周 靜,張家榮
(1.西安近代化學(xué)研究所,陜西 西安 710065;2.氟氮化工資源高效開發(fā)與利用國家重點(diǎn)實(shí)驗(yàn)室,陜西 西安 710065)
六氮雜[3.3.3]螺槳烷的多位點(diǎn)烷基化反應(yīng)
張俊林1, 王伯周1,2, 畢福強(qiáng)1, 王錫杰1,周 靜1,張家榮1
(1.西安近代化學(xué)研究所,陜西 西安 710065;2.氟氮化工資源高效開發(fā)與利用國家重點(diǎn)實(shí)驗(yàn)室,陜西 西安 710065)
以2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷-3,7,10-三酮(PTO)為原料,通過與親電試劑發(fā)生烷基化反應(yīng),獲得了具有含能化衍生前景的六烯丙基六氮雜[3.3.3]螺槳烷、六乙氧羰甲基六氮雜[3.3.3]螺槳烷和六羧甲基六氮雜[3.3.3]螺槳烷;系統(tǒng)研究了不同親電試劑與六氮雜[3.3.3]螺槳烷之間的反應(yīng)活性,探討了不同取代基六氮雜[3.3.3]螺槳烷化合物的酸堿穩(wěn)定性和熱穩(wěn)定性。結(jié)果表明,不同取代基結(jié)構(gòu)對(duì)于六氮雜[3.3.3]螺槳烷的骨架修飾具有顯著影響,親電試劑活性的增加和溶劑極性的增大對(duì)反應(yīng)有利,但過高活性的親電試劑因副反應(yīng)過多無法獲得相應(yīng)的烷基化產(chǎn)物;烷基化取代后的六氮雜[3.3.3]螺槳烷體系的水解穩(wěn)定性大大增加,酸性條件下可保持穩(wěn)定而堿性條件下多數(shù)烷基化產(chǎn)物發(fā)生降解;烷基化取代的產(chǎn)物其熱穩(wěn)定性較PTO有所增強(qiáng)。
六氮雜[3.3.3]螺槳烷;空間位阻;多位點(diǎn);烷基化;三維立體骨架
引 言
具有緊密構(gòu)型的三維立體骨架化合物往往具有較大的環(huán)應(yīng)變力及分子密度,是含能材料的理想母體結(jié)構(gòu)[1-4],從傳統(tǒng)二維平面骨架向三維立體骨架發(fā)展是提升含能化合物能量密度水平的高效途徑[5-6]。六硝基六氮雜異伍茲烷(CL-20)[7-10]及八硝基立方烷(ONC)[11-12]等分子的綜合性能優(yōu)異,是三維立體骨架型含能化合物的典型代表。氮雜[3.3.3]螺槳烷是一類含有連續(xù)季碳中心的緊湊空間結(jié)構(gòu),同時(shí)具有較大的環(huán)張力,是理想的三維立體含能化合物母體骨架[13]。2014年,Shreeve團(tuán)隊(duì)首次報(bào)道了多硝基五氮雜[3.3.3]螺槳烷含能化合物的理論研究結(jié)果,其表明氮雜[3.3.3]螺槳烷含能化合物可能成為重要的高能量密度含能化合物[14],之后Young團(tuán)隊(duì)首次利用五氮雜[3.3.3]螺槳烷中裸露的氮原子通過烷基化反應(yīng)獲得了含芐基和烯丙基的五氮雜[3.3.3]螺槳烷骨架[15]。
雖然通過縮合反應(yīng)構(gòu)建緊密三維立體骨架(如六芐基六氮雜異伍茲烷體系)具有較高的反應(yīng)效率,但通過縮合反應(yīng)實(shí)現(xiàn)該類骨架種類單一,繼CL-20之后鮮有通過縮合反應(yīng)實(shí)現(xiàn)多氮雜緊密三維立體含能化合物骨架構(gòu)建的報(bào)道,限制了三維立體含能化合物的研究,因而通過非縮合方式實(shí)現(xiàn)三維立體骨架的構(gòu)建成為含能材料研究人員面臨的巨大挑戰(zhàn)[16]。烷基化反應(yīng)對(duì)于非縮合方式構(gòu)建的多氮雜三維立體骨架的研究具有極其重要的意義,與五氮雜[3.3.3]螺槳烷相比,六氮雜[3.3.3]螺槳烷體系的空間位阻大大增加,在具有更大環(huán)張力的同時(shí)其烷基化反應(yīng)難度也隨之明顯增大,但國內(nèi)外尚無有關(guān)六氮雜[3.3.3]螺槳烷多位點(diǎn)同時(shí)烷基化的研究報(bào)道。
本研究以六氮雜[3.3.3]螺槳烷為母體進(jìn)行不同條件下的烷基化取代反應(yīng),通過控制反應(yīng)條件,選擇性地對(duì)乙氧羰甲基進(jìn)行了堿性水解而保持脲環(huán)體系不開環(huán),獲得了六羧甲基六氮雜[3.3.3]螺槳烷,為多氮雜[3.3.3]螺槳烷的研究提供參考。
1 實(shí) 驗(yàn)
1.1 試劑與儀器
烯丙基氯、烯丙基溴、氯乙酸乙酯、溴乙酸乙酯、氯化芐、溴化芐、氯丙酮、溴丙酮、溴代硝基甲烷,分析純,上海邁瑞爾試劑公司;溴甲烷、碘甲烷,分析純,北京偶合試劑公司;溴乙醛縮二乙醇、N,N-二甲基甲酰胺(DMF,超干溶劑)、二甲基亞砜(DMSO,超干溶劑),分析純,北京百靈威試劑公司;氫化鈉(質(zhì)量分?jǐn)?shù)60%),成都市科龍化工試劑廠;2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷-3,7,10-三酮(PTO),自制。
NEXUS87型傅里葉變換紅外光譜儀,美國Thermo Nicolet公司;AV 500型(500 MHz)超導(dǎo)核磁共振儀,瑞士Bruker公司;SYNAPT型UPLC-Q-TOFMS液質(zhì)聯(lián)用儀,美國Waters公司。
1.2 實(shí)驗(yàn)過程
1.2.1 合成路線
2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷(PTO)在強(qiáng)堿性條件下與不同親電試劑進(jìn)行烷基化反應(yīng),獲得不同取代基結(jié)構(gòu)的烷基化產(chǎn)物。具體合成路線如下:

1.2.2 2,4,6,8,9,11-六甲基-2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷(化合物1)的合成
0℃下,將1.98g(10.0mmol)PTO溶解于50mL DMF,冰浴冷卻至0℃,分批加入2.4g(60.0mmol)氫化鈉,0℃反應(yīng)0.5h;緩慢滴加碘甲烷8.46g(60.0mmol),撤去冰浴,室溫下(25℃)反應(yīng)3h。滴加冰水淬滅反應(yīng),將體系導(dǎo)入分液漏斗中,乙酸乙酯(50mL)萃取3次,無水硫酸鎂進(jìn)行干燥后減壓旋蒸去除溶劑,進(jìn)行柱色譜分離(乙酸乙酯與石油醚體積比1∶3)得到固體1.24g,收率44%。
1H NMR (DMSO-d6, 500 MHz),δ: 3.09 (s, 18H);13C NMR (DMSO-d6, 125MHz),δ:158.1, 88.3, 27.3; IR (KBr),ν(cm-1): 3569, 2924, 1717, 1507, 1459, 1414, 1348, 1289, 1106, 1041, 886, 871, 759, 578; MS,m/z(%): 282。
1.2.3 2,4,6,8,9,11-六芐基-2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷(化合物2)的合成
0℃下,將1.98g(10.0mmol)PTO溶解于50mL DMSO,冰浴冷卻至0℃,分批加入2.4g(60.0mmol)氫化鈉,0℃反應(yīng)0.5h;緩慢滴加溴化芐10.19g(60.0mmol),撤去冰浴,室溫下(25℃)反應(yīng)3h。滴加冰水淬滅反應(yīng),將體系導(dǎo)入分液漏斗中,乙酸乙酯(50mL)萃取3次,無水硫酸鎂進(jìn)行干燥后減壓旋蒸去除溶劑,旋蒸剩余體系加入乙醚(5mL),析出白色固體,抽濾干燥得固體2.12g,收率29%。
1H NMR (DMSO-d6, 500MHz),δ: 7.29~7.23 (m,18H), 7.11~7.09 (m, 12H), 4.32 (s, 12H);13C NMR (DMSO-d6, 125MHz),δ:157.1, 136.7, 128.6, 127.6, 126.4, 89.3, 45.6; IR (KBr),ν(cm-1): 3088, 3062, 2967, 2929, 1718,1471, 1452, 1425, 1333, 1142, 1075; MS,m/z(%): 738。
1.2.3 2,4,6,8,9,11-六烯丙基-2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷(化合物3)的合成
0℃下,將1.98g(10.0mmol)PTO溶解于50mL二甲基亞砜中,冰浴冷卻至0℃,分批加入2.4g(60.0mmol)氫化鈉,0℃反應(yīng)0.5h;緩慢滴加烯丙基溴7.2g(60.0mmol),撤去冰浴,室溫下(25℃)反應(yīng)3h。滴加冰水淬滅反應(yīng),將體系導(dǎo)入分液漏斗中,乙酸乙酯(50mL)萃取3次,無水硫酸鎂進(jìn)行干燥后減壓旋蒸去除溶劑,旋蒸剩余體系加入乙醚(5mL),析出白色固體,抽濾干燥得固體2.58g,收率61%。
1H NMR(CDCl3, 500MHz),δ: 5.80~5.72 (6H, m), 5.14~5.19 (12H, m),4.01 (12H, d, J=5.2 Hz);13CNMR (CDCl3, 500MHz, 125 MHz),δ: 156.5, 133.2, 117.2, 88.1, 44.5;IR (KBr),ν(cm-1): 3048, 2985, 2924, 1709, 1639, 1481,1421, 1334, 1295, 1223, 1152, 1101, 991, 950, 926, 857, 744; MS,m/z(%): 438。
1.2.4 2,4,6,8,9,11-六乙氧羰甲基-2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷(化合物4)的合成
0℃下,將1.98g(10.0mmol)PTO溶解于50mL二甲基亞砜中,冰浴冷卻至0℃,分批加入2.4g(60.0mmol)氫化鈉,0℃反應(yīng)0.5h;緩慢滴加溴乙酸乙酯9.9g(60.0mmol),撤去冰浴,室溫下(25℃)反應(yīng)3h。滴加冰水淬滅反應(yīng),將體系導(dǎo)入分液漏斗中,乙酸乙酯(5mL)萃取3次,無水硫酸鎂進(jìn)行干燥后減壓旋蒸去除溶劑,進(jìn)行柱色譜分離(乙酸乙酯∶石油醚/1∶2)得黃棕色油狀液體3.8g,產(chǎn)率54%。
1H NMR(CDCl3, 500MHz),δ: 4.26 (12H, s), 4.18 (12H, q,J=7.05Hz), 1.26 (18H, t,J=7.05Hz);13CNMR(CDCl3, 500MHz, 125MHz),δ: 169.5, 159.7, 90.3, 61.6, 44.1, 14.0;IR (KBr),ν(cm-1): 2985, 2940, 1739, 1455, 1376, 1299, 1209, 1026, 974, 944, 857, 753; MS,m/z(%): 714。
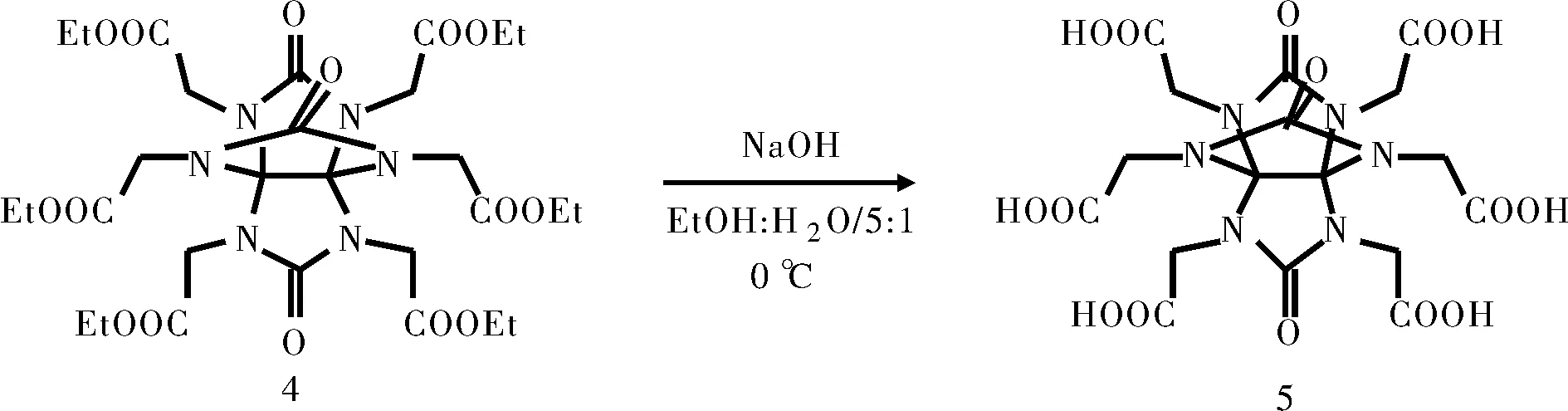
1.2.5 2,4,6,8,9,11-六羧甲基-2,4,6,8,9,11-六氮雜[3.3.3]螺槳烷(化合物5)的合成
0℃下,將719mg(1.0mmol)化合物4溶解于30mL乙醇中,冰浴冷卻至0℃,將6mL濃度為1mol/L的NaOH溶液冷卻至10 ℃左右,緩慢滴入化合物4的乙醇溶液中,加完后0℃反應(yīng);TLC實(shí)時(shí)監(jiān)測(cè),待原料消耗完后立即滴加稀鹽酸調(diào)節(jié)pH值為5~6,將反應(yīng)體系直接進(jìn)行減壓蒸餾除去溶劑得絮狀固體產(chǎn)物335mg,收率62 %。
1H NMR(CDCl3, 500MHz),δ: 4.70 (12 H, s), 3.92 (6 H, s);13C NMR (CDCl3, 500MHz,125MHz),δ: 175.0, 158.3, 88.3, 46.0;IR (KBr),ν(cm-1): 3418, 1713, 1598, 1506, 1394, 1313, 1152, 975, 658; MS,m/z(%): 546。
2 結(jié)果與討論
2.1 親電試劑和溶劑對(duì)烷基化反應(yīng)的影響
實(shí)驗(yàn)結(jié)果表明,PTO與親電試劑的烷基化反應(yīng)僅在DMF與DMSO溶劑中發(fā)生,且溶劑的選擇對(duì)反應(yīng)收率影響明顯。表1為溶劑和親電試劑對(duì)烷基化反應(yīng)的影響。

表1 溶劑和親電試劑對(duì)烷基化反應(yīng)的影響
由表1可知,碘甲烷(MeI)與PTO的烷基化反應(yīng)在DMF溶劑體系下能夠順利實(shí)現(xiàn)轉(zhuǎn)化,在DMSO中收率急劇降低。烯丙基溴、溴乙酸乙酯與PTO的烷基化反應(yīng)在DMF中無法實(shí)現(xiàn),在DMSO中轉(zhuǎn)化率則較高。芐溴(BnBr)與PTO的烷基化反應(yīng)收率在DMSO中比DMF中略高。
堿作用下氮負(fù)離子與鹵代物反應(yīng)均以雙分子親核取代反應(yīng)(SN2)為主,而DMF(偶極矩3.82)與DMSO(偶極矩3.96)兩者偶極矩差距并不明顯,但溶劑選擇對(duì)PTO烷基化反應(yīng)收率影響較大,原因尚不完全明確。此外,碘甲烷的甲基化反應(yīng)在 DMSO中基本上得不到目標(biāo)產(chǎn)物,可能的原因是碘甲烷與DMSO反應(yīng)生成锍鹽,在堿存在下生成了硫葉立德[17]。
芐基鹵素、烯丙基鹵素、乙氧羰甲基鹵素等有利于形成共軛正離子,因而反應(yīng)活性較高,此外,鹵化物反應(yīng)性表明:碘代物活性>溴代物活性?氯代物活性。從表1數(shù)據(jù)可知,對(duì)于甲基鹵素等活性較差的親電試劑,需要利用活性最高的碘代物方能發(fā)生相應(yīng)轉(zhuǎn)化,溴代物等無法實(shí)現(xiàn)轉(zhuǎn)化;芐基鹵素、烯丙基鹵素和乙氧羰甲基鹵素中利用溴化物即能夠?qū)崿F(xiàn)烷基化反應(yīng),而氯化芐不反應(yīng)。丙酮基溴和硝酸乙烷溴鹵素?zé)o法獲得相應(yīng)烷基化產(chǎn)物,可能的原因是對(duì)于該類試劑,兩個(gè)吸電子官能團(tuán)的存在造成中間的亞甲基酸性很強(qiáng),PTO與氫化鈉脫氫后形成的氨基鈉是強(qiáng)堿,在氨基鈉作用下造成亞甲基堿性下被拔氫使得無法實(shí)現(xiàn)預(yù)期的轉(zhuǎn)化過程。
2.2 烷基化取代對(duì)六氮雜[3.3.3]螺槳烷體系穩(wěn)定性的影響
2.2.1 對(duì)水解穩(wěn)定性的影響
對(duì)于未進(jìn)行烷基化取代反應(yīng)的PTO,在較強(qiáng)的酸性條件下,化合物迅速發(fā)生降解反應(yīng),表明脲羰基在氮原子無保護(hù)的情況下極易水解導(dǎo)致開環(huán)。相反,如果將PTO進(jìn)行烷基化保護(hù),則其穩(wěn)定性將大大增強(qiáng)。表2為化合物1~5及PTO在酸性條件下的穩(wěn)定性對(duì)比。對(duì)比化合物3與PTO在酸性條件下的穩(wěn)定性可知,化合物3在質(zhì)量分?jǐn)?shù)37%的濃鹽酸體系加熱條件下依然保持穩(wěn)定。化合物1、2、4、5也表現(xiàn)出類似的特性,表明烷基化取代反應(yīng)后,六氮雜[3.3.3]螺槳烷環(huán)系具有相對(duì)較強(qiáng)的水解穩(wěn)定性。

表2 化合物1~5及PTO在酸性條件下的穩(wěn)定性
在強(qiáng)堿性條件下,PTO及烷基化取代的六氮雜[3.3.3]螺槳烷環(huán)系在1mol/L NaOH中均完全降解,表明脲環(huán)結(jié)構(gòu)在堿性條件下容易發(fā)生水解反應(yīng)。由于酯基在堿性條件下的皂化反應(yīng)速率快于脲羰基的堿性水解反應(yīng),故化合物4有可能通過控制反應(yīng)條件實(shí)現(xiàn)選擇性的酯基水解并保持脲羰基結(jié)構(gòu)不變。通過條件篩選,發(fā)現(xiàn)乙醇存在的條件下,通過降低反應(yīng)溫度并進(jìn)行實(shí)時(shí)監(jiān)測(cè),在原料消耗完全之后立刻加入1mol/L鹽酸進(jìn)行中和,發(fā)現(xiàn)化合物4優(yōu)先發(fā)生酯基水解反應(yīng),獲得羧基結(jié)構(gòu)5,收率62%。
2.2.2 對(duì)熱穩(wěn)定性的影響
圖1為PTO及化合物1~5的DSC曲線。

圖1 PTO及化合物1~5的DSC曲線Fig.1 DSC curves of PTO and compounds 1-5
六氮雜[3.3.3]螺槳烷將3組五元環(huán)通過碳碳鍵進(jìn)行連接,形成連續(xù)的四級(jí)碳中心,高度緊湊的結(jié)構(gòu)迫使環(huán)系之間存在較大排斥力,從而降低了環(huán)系的穩(wěn)定性,因而六氮雜[3.3.3]螺槳烷類化合物的熱穩(wěn)定性較差。由圖1可知,PTO在溫度升高過程中全程緩慢發(fā)生熱分解,無明顯的強(qiáng)放熱峰,表明PTO在加熱過程中較易發(fā)生鍵的斷裂。與不同結(jié)構(gòu)的親電試劑發(fā)生烷基化反應(yīng)之后所形成的多取代六氮雜[3.3.3]螺槳烷體系,熱穩(wěn)定性有所增強(qiáng),多數(shù)在300℃以下穩(wěn)定性較好。
3 結(jié) 論
(1)以PTO為原料,通過親電試劑發(fā)生烷基化反應(yīng),首次獲得了具有含能化衍生前景的六烯丙基六氮雜[3.3.3]螺槳烷、六乙氧羰甲基六氮雜[3.3.3]螺槳烷和六羧甲基六氮雜[3.3.3]螺槳烷。
(2)探討了溶劑效應(yīng)及親電試劑對(duì)大位阻六氮雜[3.3.3]螺槳烷多位點(diǎn)同時(shí)烷基化的影響,表明親電試劑活性的增加和溶劑極性的增大對(duì)反應(yīng)有利,過高活性的親電試劑因副反應(yīng)過多無法獲得相應(yīng)的烷基化產(chǎn)物。
(3)烷基化取代后的六氮雜[3.3.3]螺槳烷體系的水解穩(wěn)定性大大增加,在強(qiáng)酸性條件下依然保持穩(wěn)定;堿性條件下,多數(shù)烷基化產(chǎn)物無法穩(wěn)定存在,表明大張力脲環(huán)羰基結(jié)構(gòu)在堿性條件下易發(fā)生水解反應(yīng),但通過控制反應(yīng)條件,可以實(shí)現(xiàn)酯基結(jié)構(gòu)的選擇性皂化反應(yīng)。烷基化取代的產(chǎn)物其熱穩(wěn)定性較PTO有所增強(qiáng)。
[1] 邢恩會(huì), 米鎮(zhèn)濤, 張香文. 用作新型高密度燃料的高張力籠狀烴的研究進(jìn)展[J]. 火炸藥學(xué)報(bào), 2004, 27(2): 13-16. XING En-hui, MI Zhen-tao, ZHANG Xiang-wen, et al. Development of high strained caged hydrocarbons used as high density fuels[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao),2004,27(2):13-16.
[2] Tan B, Long X, Li J, et al. The cage strain energies of high-energy compounds[J]. Computational & Theoretical Chemistry,2012,993(8): 66-72.
[3] Zhao G, Lu M. A theoretical investigation of a potential high energy density compound 3,6,7,8-tetranitro-3,6,7,8-tetraaza -tricyclo[3.1.1.1(2,4)]octane[J]. Química Nova,2013, 36(4): 513-518.
[4] Zhang J Y, Wang F, Gong X D. A DFT study of cage compounds: 3, 5, 8, 10, 11, 12-hexanitro-3, 5, 8, 10, 11, 12-hexaazatetracyclo [5.5.1.12,6.04,9] dodecane and its derivatives as high energetic materials[J]. Structural Chemistry, 2013, 24(4):1339-1346.
[5] Ju X, Wang Z. Prediction of caged polyaza polynitroamine (tetracyclo-HMX) as energetic compound[J]. Journal of Energetic Materials,2009, 27(2): 133-143.
[6] Zhang Q, Shreeve J M. Metal-organic frameworks as high cxplosives: a new concept for energetic materials[J]. Angewandte Chemie International Edition,2014, 53(10): 2540-2542.
[7] Nielsen A T. Caged polynitramine compound: US, Patent 5,693,794[P]. 1997-12-2.
[8] Nair U R, Sivabalan R, Gore G M, et al. Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-based formulations (review)[J]. Combustion Explosion & Shock Waves,2005, 41(2):121-132.
[9] 宋小蘭,王毅,宋朝陽,等.CL-20/DNT共晶炸藥的制備及其性能研究[J].火炸藥學(xué)報(bào),2016,39(1):23-27. SONG Xiao-lan, WANG Yi, SONG Zhao-yang, et al. Preparation of CL-20/DNT cocrystal explosive and study on its performance[J]. Chinese Journal of Explosives & Propellants (Huozhayao Xuebao), 2016,39(1):23-27.
[10] 高寒,劉杰,郝嘎子,等.納米CL-20的制備、表征和粉碎機(jī)理研究[J].火炸藥學(xué)報(bào),2015,38(2):46-49. GAO Han, LIU Jie, HAO Ga-zi, et al.Study on preparation, characterization and comminution mechanism of nano-sized CL-20[J]. Chinese Journal of Explosives & Propellants (Huozhayao Xuebao), 2015,38(2):46-49.
[11] Zhang M X, Eaton P E, Gilardi R. Hepta- and octanitrocubanes[J]. Angewandte Chemie International Edition,2000, 39(2):401-404.
[12] Hrovat D A, Borden W T, Eaton P E, et al. A computational study of the interactions among the nitro groups in octanitrocubane[J]. Journal of the American Chemical Society,2001, 123(7):1289-1293.
[13] 張俊林, 肖川, 翟連杰, 等. 多硝基氮雜稠環(huán)化合物的合成及性能, 有機(jī)化學(xué)[J], 2016, 36(6): 1197-1207. ZHANG Jun-lin, XIAO Chuan, ZHAI Lian-jie, et al. Synthesis and properties of the fused aza-polynitrocyclic compounds[J]. Chinese Journal of Organic Chemistry,2016, 36(6): 1197-1207.
[14] Zhang Q, Zhang J, Qi X, et al. Molecular design and property prediction of high density polynitro[3.3.3]-propellane-derivatized frameworks as potential high explosives[J]. Journal of Physical Chemistry A, 2014, 118(45):10857-10865.
[15] Shin M, Kim M H, Ha T H, et al. Synthesis of novel 2,4,6,8,10-pentaaza[3.3.3]propellane derivatives[J]. Tetrahedron, 2014, 70(8):1617-1620.
[16] Agrawal J P, Hodgson R D. Organic Chemistry of Explosives[M]. West Sussex:John Wiley & Sons, Ltd.,2007.
[17] Corey E J, Chaykovsky M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and dimethylsulfonium methylide ((CH3)2SCH2).Formation and application to organic synthesis[J]. Journal of the American Chemical Society, 2002, 87(6):1353-1364.
Multiple Site N-Alkylation Reactivity of Hexaaza[3.3.3]propellane
ZHANG Jun-lin1, WANG Bo-zhou1,2, BI Fu-qiang1, WANG Xi-jie1, ZHOU Jing1, ZHANG Jia-rong1
(1.Xi′an Modern Chemistry Research Institute, Xi′an 710065, China;2.State Key Laboratory of Fluorine& Nitrogen Chemicals, Xi′an 710065, China)
Taking 2,4,6,8,9,11-hexaaza[3.3.3]propellanes-3,7,10-trione (PTO) as raw material, the reactivity of the hexaaza[3.3.3]propellane with different electrophilic reagents was systematically investigated. N-hexallyl-hexaaza[3.3.3] propellanes, N-hexethylacetic -hexaaza[3.3.3] propellanes and N-hexacetoxyl-hexaaza[3.3.3] propellanes with energetic derivative prospect were designed and synthesized for the first time. The acid-stability, base-stability and thermal stability of hexaaza[3.3.3]propellane with different substituent were discussed. The results show that different substituent structure has significant effect on the modification of the hexaaza[3.3.3]propellane skeleton.Increasing the activity of electrophilic reagent and solvent polarity enhanced the reaction process, but extremely high activity failed to obtain the corresponding alkylation products due to the adverse reactions. The hydrolytic stability of the N-alkylated hexaaza[3.3.3]propellane system is greatly increased. Most of them remained stable under acidic conditions while decomposed under alkaline conditions. The thermal stability of the products was enhanced by alkylation compared with PTO.
hexaaza[3.3.3]propellane; steric hindrance; multiple reactive sites; alkylation; three-dimensional skeleton
10.14077/j.issn.1007-7812.2017.04.006
2016-09-16;
2017-04-29
國家自然科學(xué)基金(No.21503162)
張俊林(1986-),男,博士,從事含能材料合成研究。E-mail: junlin-111@163.com
畢福強(qiáng)(1982-),男,博士,高級(jí)工程師,從事含能材料合成研究。E-mail: bifuqiang@msn.com
TJ55;O621
A
1007-7812(2017)04-0033-05
