MIP-SERS技術(shù)快速檢測中藥中非法添加的茶堿
2017-08-01 12:46:37卞筱泓許激揚
分析測試學報 2017年7期
胡 然,陸 峰,卞筱泓,許激揚*
(1.中國藥科大學 生命科學與技術(shù)學院,江蘇 南京 211198;2.第二軍醫(yī)大學 藥學院,上海 200433)
?
MIP-SERS技術(shù)快速檢測中藥中非法添加的茶堿
胡 然1,陸 峰2,卞筱泓1,許激揚1*
(1.中國藥科大學 生命科學與技術(shù)學院,江蘇 南京 211198;2.第二軍醫(yī)大學 藥學院,上海 200433)
建立了分子印跡技術(shù)(MIP)與表面增強拉曼光譜(SERS)聯(lián)用對中藥中非法添加的茶堿成分快速檢測的方法。基于沉淀聚合方法合成MIP微球,對復雜中藥基質(zhì)中的茶堿進行簡單分離,采用SERS對MIP中吸附的茶堿進行定性檢測。實驗利用SEM,F(xiàn)T-IR對MIP結(jié)構(gòu)表征,考察了MIP的熱力學、動力學和選擇性吸附能力,以及摻雜成分的檢出限。結(jié)果表明,MIP比NIP對茶堿具有更好的特異性吸附和選擇性,該方法對茶堿的檢出限低至0.1 μg/L。方法用于5種止咳平喘類中藥的檢測,其中1種中藥檢出非法添加了茶堿成分。該法靈敏度高,特異性強,無需前處理,簡單快速,實現(xiàn)了摻偽中藥中茶堿的快速檢測,有望進一步應用于其他復雜基質(zhì)體系。
分子印跡技術(shù);表面增強拉曼光譜;中藥摻偽;茶堿
近年來,中藥及保健品中非法添加化學成分的現(xiàn)象屢見不鮮,愈演愈烈,2017年1月,國家食藥總局查處了近200批次的中藥飲片,檢測發(fā)現(xiàn)存在染色、增重、摻假現(xiàn)象。這些非法添加的化學品由于本身的毒性或因隨意大量添加均會對人體產(chǎn)生巨大的危害,甚至造成死亡。
目前分析領(lǐng)域中常見的檢測手段包括傳統(tǒng)的高效液相色譜法(HPLC)[1]、液相色譜-質(zhì)譜聯(lián)用法(LC-MS)[2]、氣相色譜-質(zhì)譜聯(lián)用法(GC-MS)[3]、薄層色譜-表面增強拉曼光譜法(TLC-SERS)[4]、表面等離子共振(SPR)[5]等,但這些方法具有耗時,成本高,前處理繁瑣,對儀器和操作人員要求高,需大量有機溶劑等缺點,不能滿足當前分析檢測領(lǐng)域快速、準確、簡單的要求。
分子印跡技術(shù)(Molecular impriting technology,MIT)通過“抗原-抗體”原理制備仿生材料,以目標分析物作為模板,能夠在復雜基質(zhì)中特異性識別和分離出目標分子,從而達到有效分離和富集的作用。表面增強拉曼光譜(Surface-enhanced Raman scattering,SERS)是一種活性拉曼分子在金屬表面信號被強烈放大的分子譜學技術(shù),其靈敏度高,甚至可實現(xiàn)單分子檢測。目前已有文獻報道“MIP-SERS”聯(lián)用技術(shù)在食品藥品中的檢測應用,包括檢測蘇丹紅Ⅰ[6]、三聚氰胺[7]、普萘洛爾[8]、氯霉素[9]等,但國內(nèi)的相關(guān)研究報道較少。本文采用“MIP-SERS”對中藥中非法添加茶堿進行了檢測研究,以期達到快速、簡單、特異性好和靈敏度高的目的。
1 實驗部分
1.1 儀器與試劑
XW-80A型旋渦混合器(上海精科實業(yè)有限公司);DF-101S集熱式恒溫加熱磁力攪拌器(上海梅穎浦儀器儀表制造有限公司);KQ-250DB型數(shù)控超聲波清洗器(昆山市超聲儀器有限公司);TU-1901型雙光束紫外可見分光光度計(北京普析通用儀器有限公司);TG16-WS高速離心機(上海盧湘儀離心機有限公司);BWS415-785H便攜式拉曼光譜儀(B&W Tek,USA);ALPHA傅立葉變換紅外光譜儀(德國Bruker公司);掃描電鏡(ZeissEVOMA-10)。
甲基丙烯酸(MAA)、二甲基丙烯酸乙二醇酯(EGDMA)、偶氮異二丁腈(AIBN)均購自阿拉丁試劑有限公司,使用前均去除阻聚劑;茶堿、咖啡因、多索茶堿均由中國食品藥品檢定研究院提供;甲醇、乙腈、冰乙酸等購于國藥集團化學試劑有限公司,除乙腈為色譜純外,其他均為分析純;中藥樣品由山東省食品藥品檢驗所提供。實驗用水為去離子水。
1.2 茶堿MIP的合成
茶堿MIP的合成方法主要參考文獻[10],取茶堿47 mg,加入40 mL乙腈,超聲溶解,依次加入87.5 μL MAA,770 μL EGDMA以及14 mg AIBN,氮吹10 min,密封,60 ℃水浴聚合6 h,將聚合物離心,甲醇洗去多余乙腈,再用甲醇-冰乙酸(8∶2,體積比)振蕩洗脫,直至紫外檢測不到茶堿模板分子,最后用甲醇洗去殘余冰乙酸,用水洗去甲醇,真空干燥后即得MIP。非印跡聚合物除了不加茶堿外,其余操作步驟同上。
1.3 納米銀膠的合成
根據(jù)文獻“Lee”法膠[11]的合成方法,將36 mg AgNO3溶于10 mL水,置于三頸燒瓶中,加入190 mL水加熱至微沸,同時將100 mg檸檬酸鈉溶于10 mL水,取4 mL快速加入三頸燒瓶中,加熱回流40~50 min即可。
1.4 吸附能力考察
配制不同濃度的茶堿標準水溶液,建立標準曲線,分別取2 mL茶堿標準溶液與5 mg茶堿MIP和茶堿NIP在25 ℃室溫下振蕩12 h,離心,取上清液過0.22 μm濾膜,測其紫外吸光度。取一定濃度的茶堿水溶液,加入5 mg茶堿MIP,在10~180 min后分別測量吸附后的紫外吸光度。
1.5 MIP的拉曼表征
將結(jié)構(gòu)相似的10 mg/L的咖啡因、茶堿、多索茶堿溶液分別與5 mg MIP微球混合,振蕩12 h后,與等體積的銀膠1∶1混合后采集拉曼信號。 配制不同濃度的茶堿水溶液,分別與5 mg MIP材料混合振蕩2 h后,與等體積的銀膠1∶1混合后采集拉曼信號。
1.6 中藥樣品的檢測
取5份中藥真實樣品適量,研細,加入水中超聲5 min,離心取上清液與5 mg MIP材料振蕩10 min,離心,水洗3次,與銀膠1∶1混合后采集其拉曼信號。
2 結(jié)果與討論
2.1 納米銀膠的表征
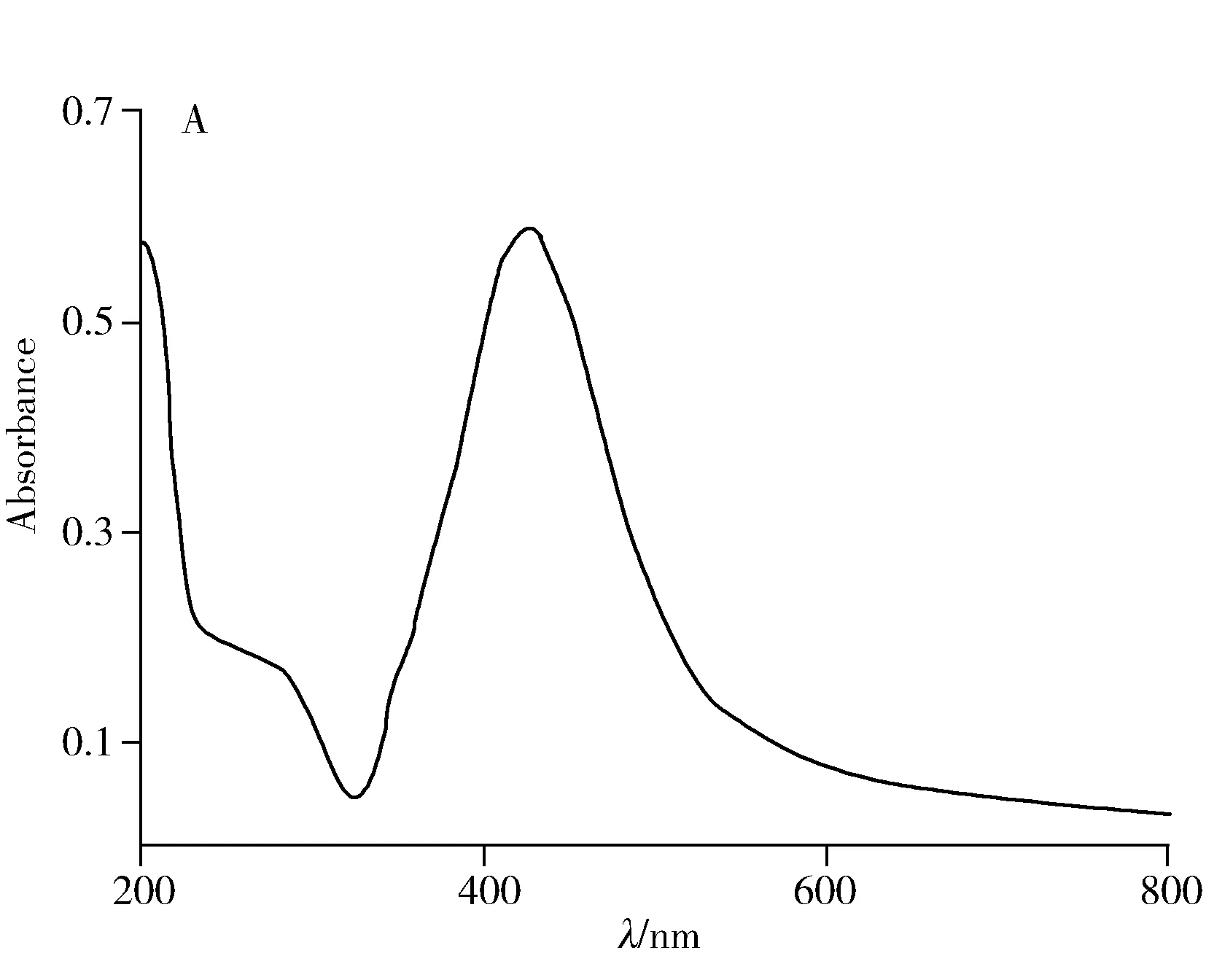
本文選擇最常用的Lee法膠作為銀溶膠,通過紫外圖譜可以觀察到納米銀在420 nm左右有1個較強的單峰(圖1A),反映出納米銀顆粒呈較好的均一性球形,其直徑約為40~60 nm[12],進一步通過掃描電鏡(圖1B)佐證納米銀顆粒的形狀和直徑,為SERS的增強效果和穩(wěn)定性提供了保證。
2.2 茶堿MIP的合成與表征
2.2.1 茶堿與功能單體比例的優(yōu)化 采用紫外光譜對茶堿模板分子和甲基丙烯酸功能單體的比例(茶堿空白,1∶1、1∶2、1∶4、1∶6、1∶8)進行篩選。觀察到當甲基丙烯酸的比例不斷增大時,最大吸收波長從275 nm紅移至277 nm,這主要是由于茶堿與甲基丙烯酸的羧基發(fā)生了氫鍵結(jié)合[13],當模板分子和功能單體的比例為1∶4時,繼續(xù)增大甲基丙烯酸的比例,最大吸收波長基本不發(fā)生變化,說明1個茶堿分子可結(jié)合4個甲基丙烯酸。
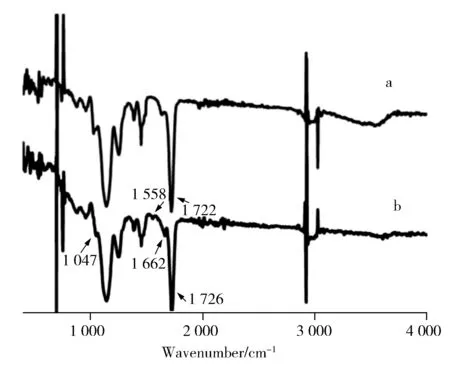
圖2 MIP(a)和NIP(b)吸附茶堿后的紅外光譜圖Fig.2 FT-IR spectra of MIP(a) and NIP(b) after adsorpting theophylline
2.2.2 茶堿MIP及茶堿NIP 的掃描電鏡圖 采用掃描電鏡對茶堿MIP及NIP進行表征。實驗結(jié)果顯示,NIP的均一性優(yōu)于MIP,可能是由于相對于NIP 微球,MIP微球內(nèi)部需要更多的茶堿孔穴,造成了大小、孔徑和分布不均一[14],這種微觀的不均一性在宏觀上呈現(xiàn)出微球大小的不均一。

2.3 MIP的吸附曲線
茶堿與MIP和NIP分別振蕩12 h后,離心,考察了MIP和NIP對茶堿的靜態(tài)結(jié)合等溫線。實驗結(jié)果顯示,MIP對茶堿的吸附能力明顯強于NIP,MIP對茶堿的飽和吸附量約為22 mg/g,而NIP不足10 mg/g。這主要是因為MIP以茶堿為模板形成的三維孔穴與茶堿分子更為接近,而NIP的孔穴卻無均一性,對茶堿的選擇性更差。因此,對于相同濃度的茶堿溶液,MIP的吸附能力好于NIP。
2.4 THO@MIP的拉曼識別特性
圖3分別為0.5 mg/L茶堿溶液與MIP和NIP結(jié)合后的SERS圖譜以及茶堿本身的SERS圖譜,從圖中可以看到571,696,1 305 cm-1處的峰主要來自茶堿嘧啶和咪唑環(huán)的伸縮振動,1 258 cm-1處屬于嘧啶環(huán)上的甲基振動峰,在吸附茶堿后,MIP的茶堿峰增大明顯好于NIP,這主要是MIP對茶堿的特異性吸附導致。
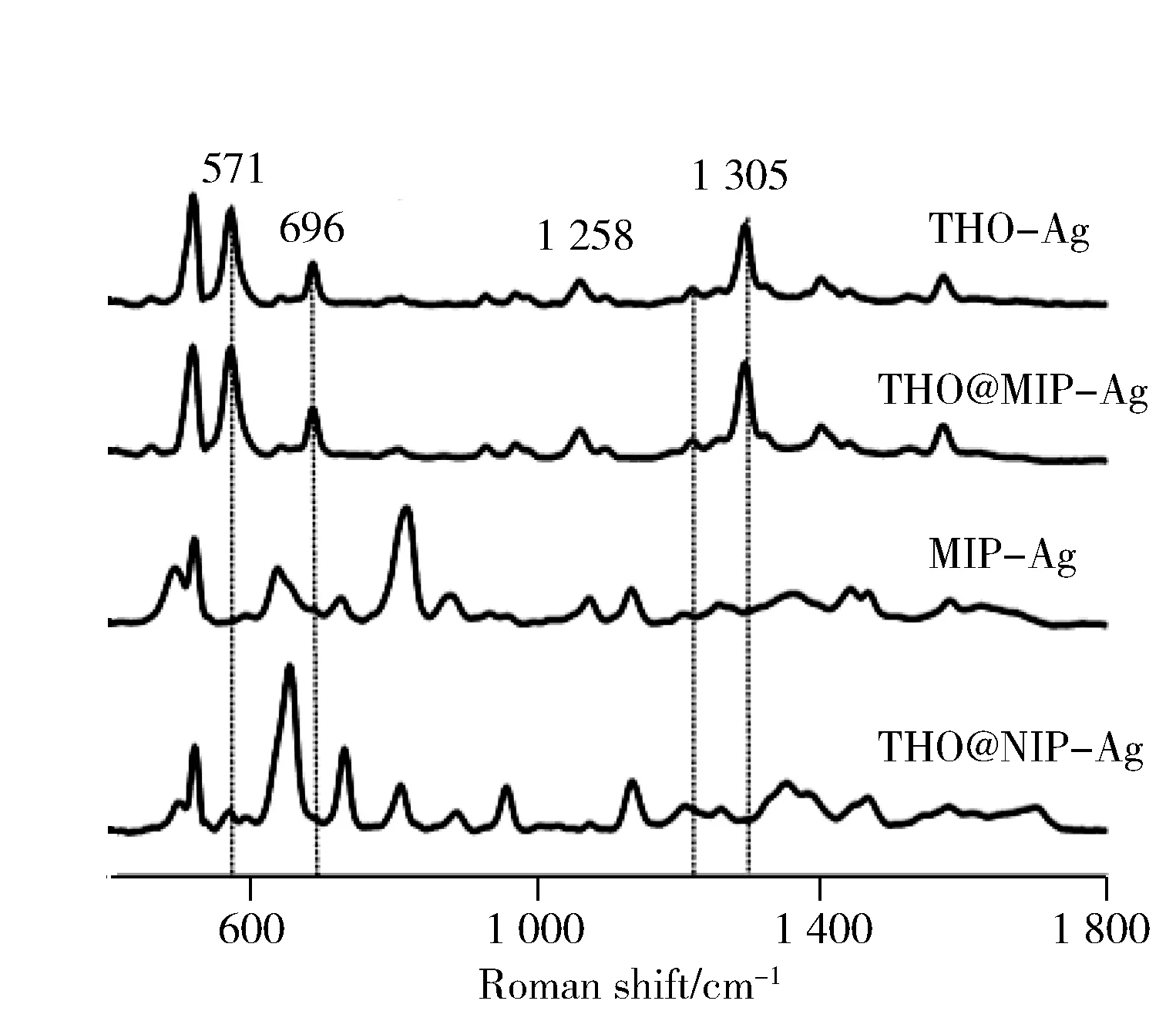
圖3 MIP和NIP結(jié)合茶堿后的SERS圖譜Fig.3 SERS spectra of MIP and NIP after rebinding of theophylline
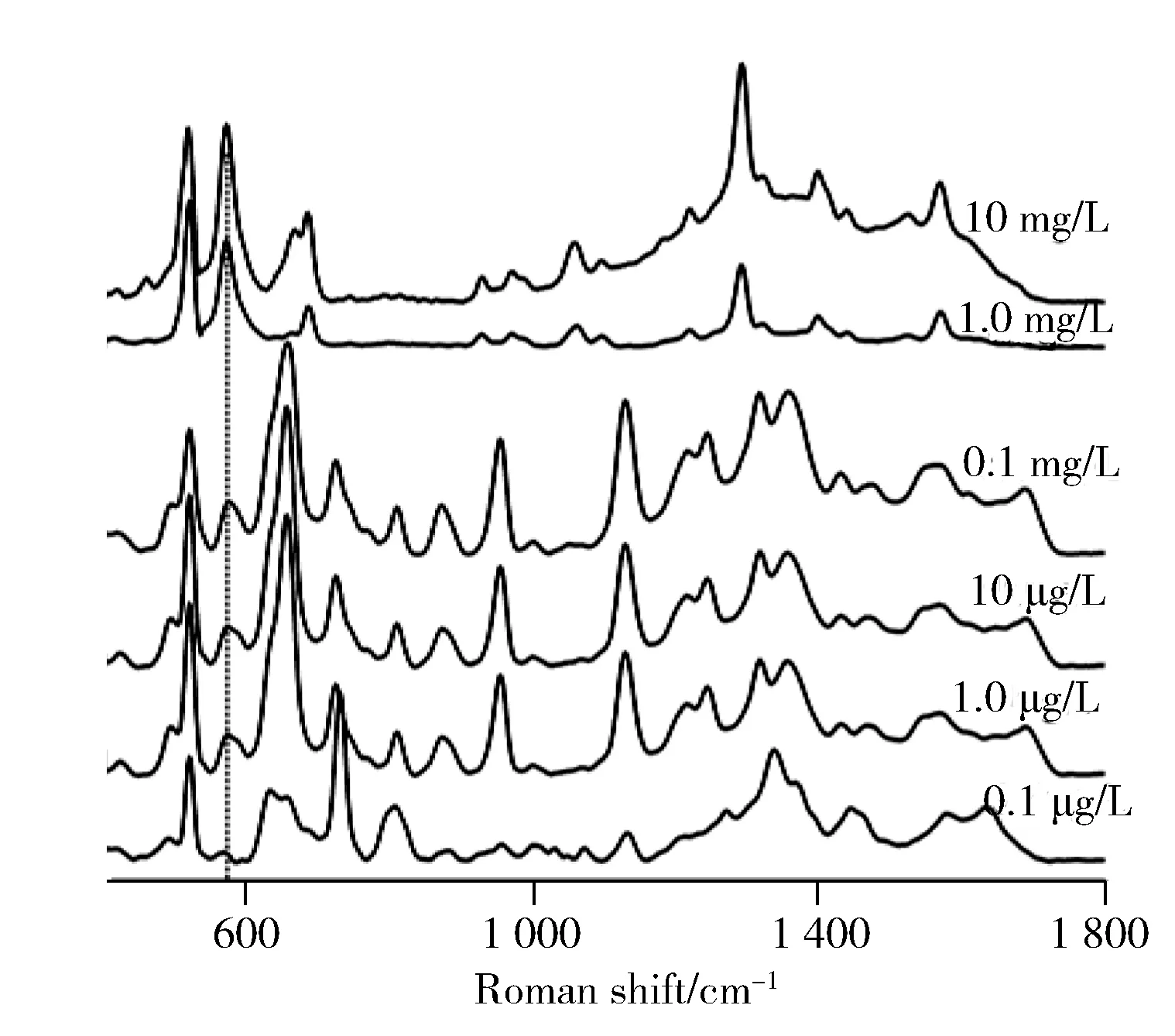
圖4 MIP-SERS對茶堿檢出限Fig.4 Detection limit of theophylline with MIP-SERS
2.5 THO@MIP的拉曼選擇性
為了進一步考察MIP的選擇性,將0.5 mg/L的茶堿、咖啡因、多索茶堿溶液分別與MIP混合攪拌一段時間。結(jié)果顯示,茶堿的SERS信號好于咖啡因和多索茶堿,這也證明在結(jié)構(gòu)類似物中,茶堿比咖啡因和多索茶堿更適合MIP中的孔穴,而相比于茶堿,咖啡因由于多1個甲基官能團,導致位阻增大,因此對MIP的孔穴識別能力下降,而多索茶堿的官能團更大,這可能是咖啡因的SERS強于多索茶堿的原因之一。
2.6 MIP-SERS檢出限
考察了不同濃度的茶堿溶液與MIP攪拌10 min后的拉曼信號。結(jié)果顯示,當茶堿濃度大于1.0 mg/L 時,拉曼圖譜以茶堿信號為主,而當濃度低于1.0 mg/L時,拉曼圖譜發(fā)生了變化(見圖4),考慮此時茶堿濃度過低,拉曼峰以MIP材料的拉曼峰為主,571 cm-1和1 305 cm-1的兩個尖銳峰屬于茶堿分子不同振動模式的峰,但1 305 cm-1處的特征峰在低濃度時干擾較大,所以選擇571 cm-1的茶堿特征峰為檢出限的識別特征峰。以571 cm-1的茶堿特征峰為標準,當茶堿濃度低于1.0 μg/L時,仍然可以判斷目標基質(zhì)中含有茶堿,低于文獻報道的10-7mol/L[16]。
2.7 實際樣品的考察
采用MIP-SERS對5份真實中藥樣品進行檢測,除了3號樣品外,其他4份樣品在571 cm-1茶堿特征峰處均未出峰,而3號樣品在571 cm-1處有強峰,表明3號樣品非法添加了茶堿。本方法整個檢測過程不超過30 min,完全滿足快速檢測的要求。
3 結(jié) 論
本文以茶堿為檢測對象,將“MIP-SERS”分析手段運用于中藥中茶堿摻偽的快速檢測,該方法靈敏度高,特異性強,簡單,快速,滿足當前分析檢測方法的發(fā)展要求。“MIP-SERS”有望進一步應用于其他分析領(lǐng)域。
[1] Fan Q Y,Hao Y H,Dai C M.J.Instrum.Anal.(樊輕亞,郝艷紅,代春美.分析測試學報),2017,36(1):106-111.
[2] Guo X,Sun J H,Sun Z Z,Liu J H,Huang X L,Liu Y L,Chen L.J.Instrum.Anal.(郭霞,孫建華,孫建中,劉菁華,黃雪玲,劉云璐,陳琳.分析測試學報),2016,35(12):1535-1541.
[3] Wang Y Y,Xu Z Q,Li D,Zheng J G,Zhou M H,Liu Y F,Xiao Q,Zhong Z G.J.Instrum.Anal.(王云玉,許志欽,李丹,鄭建國,周明輝,劉瑩峰,肖前,鐘志光.分析測試學報),2014,33(6):724-727.
[4] Zhu Q X,Cao Y B,Cao Y Y,Chai Y F,Lu F.Anal.Bioanal.Chem.,2014,406(7):1877-1884.
[5] Anand M S,Satyendra K M,Banshi D G.Mater.Res.Express,2015,2(3):035007.
[6] Gao F,Hu Y X,Chen D,Li-Chan C Y L,Grant E,Lu X N.Talanta,2015,143:344-352.
[7] Hu Y X,F(xiàn)eng S L,Gao F,Li-Chan C Y L,Grant E,Lu X N.FoodChem.,2015,176:123-129.
[8] Bompart M,Wilde Y D,Haupt K.Adv.Mater.,2010,22(21):2343-2348.
[9] Gao F,F(xiàn)eng S L,Chen Z W,Li-Chan C Y L,Grant E,Lu X N.J.FoodSci.,2014,79(12):2542-2549.
[10] Ye L,Weiss R,Mosbach K.Macromolecules,2000,33(22):8239-8245.
[11] Lee P C,Meisel D.J.Phys.Chem.,1982,86(17):3391-3395.
[12] Evanoff D D,Chumanov G.J.Phys.Chem.B,2004,108(37):13957-13962.
[13] He M Y,Ye Y F,Liu Y,Li Z Z,Jiao B N,Zeng S M,Su X S.J.Instrum.Anal.(賀美艷,葉玉鳳,劉炎,李珍柱,焦必寧,曾紹梅,蘇學素.分析測試學報),2017,36(3):325-330.
[14] Chen W,Yang X H,Yang Z S,Wang Y M,Cao J L.J.Instrum.Anal.(陳文,楊曉花,楊支帥,王以明,曹靜亮.分析測試學報),2014,34(3):271-276.
[15] Nolasco M M,Amado A M,Ribeiro-Claro P J A.ChemPhysChem.,2006,7(10):2150-2161.
[16] Kan X W,Liu T T,Zhou H,Li C.Microchim.Acta,2010,171(3):423-429.
Rapid Detection of Theophylline Illegally Mixed in Traditional Chinese Medicine by MIP-SER
SHU Ran1,LU Feng2,BIAN Xiao-hong1,XU Ji-yang1*
(1.College of Science and Technology,China Pharmceutical University,Nanjing 211198,China;2.School of
Pharmacy,The Second Military Medical University,Shanghai 200433,China)
A rapid method combining molecularly imprinted polymers with surface-enhanced Raman spectroscopy(MIPs-SERS) was established for the determination of theophyline illegally added into traditional Chinese Medicine.MIPs were synthesized by precipitation polymerization,and the theophylline was simply separated from the complex traditional Chinese medicine(TCM) substrates, then detected qualitatively by SERS.The structures of the newly synthesized MIPs were characterized by Fourier transmission infrared spectrometry(FT-IR) and scanning electron microscopy(SEM).The thermodynamics and kinetics and selective adsorption capacity of the MIPs,as well as the limit of detection for adulteration were also investigated.The results showed that MIPs had a better specificity and selectivity than NIPs,and the limit of detection was as low as 1 μg/L.Five kinds of real TCM relieving cough and asthma were tested,and one sample was detected to have theophylline components added illegally. With the advantages of high sensitivity,strong specificity,no pretreatment,simplicity and rapidness,the method could be used for the rapid detection of theophylline illegally added in TCM and further applied in other complicated matrix system.
molecularly imprinted technology;surface enhanced Raman spectroscopy;traditional Chinese medicine(TCM) adulteration;theophylline
2017-02-15;
2017-04-15
國家重大儀器開發(fā)專項項目(2012YQ18013203)
10.3969/j.issn.1004-4957.2017.07.011
O657.3;TQ460.72
A
1004-4957(2017)07-0902-05
*通訊作者:許激揚,博士,教授,研究方向:藥物生物合成,Tel:025-83271306,E-mail:jiyangx@126.com
猜你喜歡
中老年保健(2021年4期)2021-12-01 11:19:40
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年4期)2021-08-22 07:08:32
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
金橋(2020年7期)2020-08-13 03:07:00
基層中醫(yī)藥(2020年12期)2020-07-22 06:34:38
基層中醫(yī)藥(2018年6期)2018-08-29 01:20:20
