丁酮3s里德堡態的超快光解動力學研究?
2017-08-01 00:35:16羅金龍凌豐姿李帥王艷梅張冰
物理學報 2017年2期
羅金龍 凌豐姿 李帥 王艷梅 張冰
1)(昆明學院物理科學與技術系,昆明 650214)
2)(中國科學院武漢物理與數學研究所,波譜與原子分子物理國家重點實驗室,武漢 430071)
丁酮3s里德堡態的超快光解動力學研究?
羅金龍1)凌豐姿2)李帥2)王艷梅2)張冰2)?
1)(昆明學院物理科學與技術系,昆明 650214)
2)(中國科學院武漢物理與數學研究所,波譜與原子分子物理國家重點實驗室,武漢 430071)
(2016年9月12日收到;2016年11月1日收到修改稿)
本文采用195.8 nm飛秒激光將丁酮分子激發到S2(n,3s)里德堡態,在800 nm探測光的作用下獲得時間分辨的飛行時間質譜.對實驗結果的分析表明,由于丁酮α位置具有一個甲基和一個乙基,使得Norrish I型解離反應表現出豐富的動力學特征.母體離子瞬態衰減的時間常數為(2.23±0.02)ps.丙酰基離子瞬態衰減與母體類似,只有一個為(2.15±0.02)ps的時間常數,說明丙酰基離子來自于母體的解離性電離.乙酰基離子的時間曲線擬合得到四個時間常數:τ1=(2.40±0.15)ps,τ2=(1.10±0.25)ps,τ3=(0.08±0.02)ps,τ4=(17.72±0.80)ps,分別對應于S2→S1的內轉換,S1態生成CH3CO()的初步解離,CH3CO()快速內轉換為CH3CO(),以及CH3CO()基態上的二次解離.丁酮分子α-C-C鍵的解離存在分子內振動能量再分配(IVR)與勢壘解離兩種競爭的解離通道,但在該實驗條件下,我們認為是通過分子內振動能量再分配通道發生解離的結果.
時間分辨質譜,超快動力學,泵浦-探測,光解離
1 引 言
脂肪酮作為一類重要的有機化合物,數十年來被進行了廣泛深入的研究[1-20].脂肪酮含有化學性質活潑的羰基,容易光解或與OH自由基發生反應[1,2].主要的光解過程是nπ?軌道上的電子被激發后,與羰基相鄰的C-C鍵發生解離,這類反應也稱為Norrish I型反應[3,4].隨著飛秒化學的發展,酮類分子的Norrish I型反應機理得到了進一步研究[5-12].目前對丁酮分子的Norrish I型反應研究報道相對較少[5-8],而事實上丁酮分子的α位置具有一個甲基和一個乙基,在Norrish I型反應中,能夠完全分辨分子中與羰基相鄰的兩個α-C-C鍵,對于認識酮類分子的α-C-C鍵的解離反應機理具有積極作用.
丁酮的真空紫外吸收譜顯示[13],丁酮在180-200 nm范圍內存在一個對應于第二電子激發單重態S2(n,3s)態躍遷的吸收帶.該吸收帶明顯的振動結構意味著S2(n,3s)并非直接解離態,而是存在預解離過程.研究丁酮分子S2(n,3s)激發態的弛豫動力學,有助于探明Norrish I型光解反應的斷鍵機理.我們的實驗通過飛秒時間分辨的飛行時間質譜技術[14,15]實時跟蹤了丁酮初始態和中間體的演化,研究S2態的光解動力學特征.在真空環境下,195.8 nm的飛秒激光將丁酮分子泵浦到S2態,然后用800 nm的飛秒激光作為探測光,相對于泵浦光經過一定的時間延遲后作用到S2態的丁酮分子上將其電離,形成的離子通過一個無場飛行管到達探測器.通過監測離子信號強度隨延遲時間的變化,從而獲得激發態的衰減壽命等動力學信息.
2 實 驗
實驗中使用的激光器是一套自鎖模鈦:藍寶石飛秒激光系統(Coherent Int.,美國),中心波長為800 nm,最大單脈沖能量為4.5 mJ,線寬8.6 nm,帶寬100 fs,頻率1 kHz.該激光器配置了TOPAS(traveling wave optic parametric amplifier system)系統(Coherent Int.,美國),在單脈沖能量約為1 mJ的飛秒激光泵浦下,輸出波長在240-2600 nm范圍內可調的激光.實驗時調節TOPAS產生260 nm的輸出光,與800 nm的基頻光通過BBO晶體混頻后產生195.8 nm的和頻光,將其作為泵浦光,單光能量為每脈沖0.2μJ,實驗光路如圖1所示.探測光則直接使用800 nm的基頻光,單光能量為每脈沖50μJ.泵浦光與探測光皆為豎直偏振,平行于探測器表面.泵浦光和探測光通過合束片合并后直接進入束源室作用在分子束上.泵浦和探測脈沖之間的時間延遲由計算機控制的線性精密平移臺(PI,M-126.GG1,德國)改變光路中平移量Δx的值進行精確控制.此外,根據實驗的需要,在泵浦和探測光路中加入衰減片,適當地衰減光強,不僅能夠減小背景信號,也有利于調節泵浦與探測光之間的強度比例,獲得更好的信噪比.
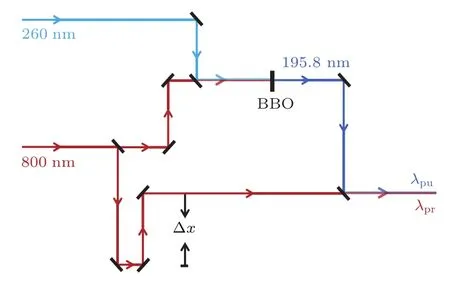
圖1 實驗光路示意圖 TOPAS產生260 nm的輸出光,與800 nm的基頻光和頻產生195.8 nm的泵浦光,探測光是800 nm的基頻光Fig.1.Diagram of the experimental pump-probe light path.The pump pulse at 195.8 nm is obtained by the sum frequency mixing of the compressed optical parametric amplifier(OPA)output at 260 nm with the fundamental pulse at 800 nm.The probe pulse is 800 nm fundamental laser.
實驗裝置包括真空系統、超聲分子束裝置、飛行時間質譜儀,和一個二維位置敏感探測器[16,17].實驗樣品分子丁酮(99%純度),使用前無需進一步純化.丁酮經氦氣攜帶通過孔徑為0.5 mm的脈沖閥(Parker,general valve,美國)絕熱膨脹產生脈沖超聲分子束.分子束經skimmer限束和準直后進入電離室,與泵浦和探測激光束垂直相交,激光作用后產生的離子通過μ金屬屏蔽的自由飛行區(360 mm),被二維位置敏感探測器接收,通過光電倍增管采集熒光屏上的光信號,輸送到示波器上獲得質譜信號,由計算機進行監控和處理.
丁酮經過超聲膨脹后,極易形成團簇[18],實驗中調節分子束的時序,使激光作用于分子束的前沿,能夠有效避免產生團簇離子.整個系統的時序控制利用一臺多通道時序控制器(DG535,stanford research systems)實現.
3 結果與討論
圖2為丁酮分子的雙光作用下的飛行時間質譜. 從質譜圖上看出,在雙光共同作用下,出現較強的母體離子(72 aum)和光解碎片離子CH3CH2CO+(57 aum),CH3CO+(43 aum)信號.如此高的雙光增益表明,195.8 nm泵浦光確實將丁酮分子激發到了3s里德堡態,與195.2 nm的理論計算值相符[19],并且所反映的都是單光子泵浦到3s態的動力學過程,因為丁酮的電離勢為9.54 eV[19],195.8 nm的能量為6.33 eV,如果是雙光子泵浦,分子就直接電離了.CH3CH2CO+和CH3CO+的出現勢分別為9.9[20]和10.32 eV[21],這意味著195.8 nm激發丁酮到S2(n,3s)里德堡態后,在不少于3個探測光子的作用下出現這些瞬態離子.
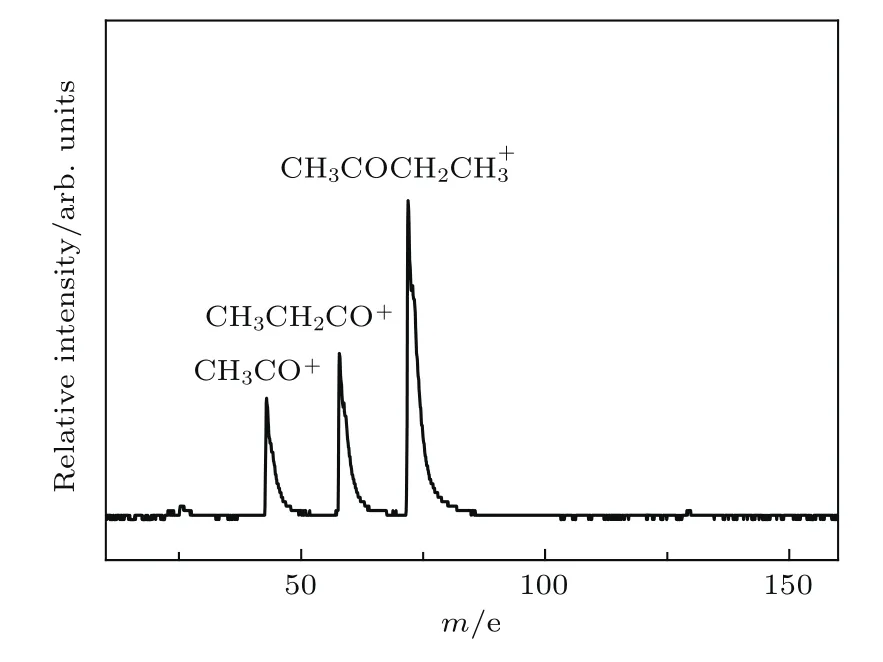
圖2 195.8和800 nm雙光作用下丁酮的飛行時間質譜Fig.2.The time-of-flight mass spectrometry(TOFMS)of butanone by 195.8 and 800 nm.
圖3給出了雙光作用下母體及典型碎片離子隨泵浦-探測時間延遲的變化曲線及擬合結果.如圖3(a)所示,母體瞬態可擬合為單指數衰減,時間常數τa=(2.23±0.02)ps,是S2態到第一電子激發單重態S1態內轉換(IC)的時間,表示丁酮S2態帶源壽命.而且,母體瞬態單一指數的衰減意味著S2→S0態內轉換以及S2→T1系間竄越(ISC)在該實驗結果的分析中可以忽略.圖3(b)中丙酰離子相對強度迅速增加,得到的時間常數τb=(2.15±0.02)ps與母體離子擬合得到的衰減時間十分接近,這意味著丙酰離子完全來自于母體離子的解離性電離,反映的是S2→S1內轉換過程,τb是內轉換所需要的時間.
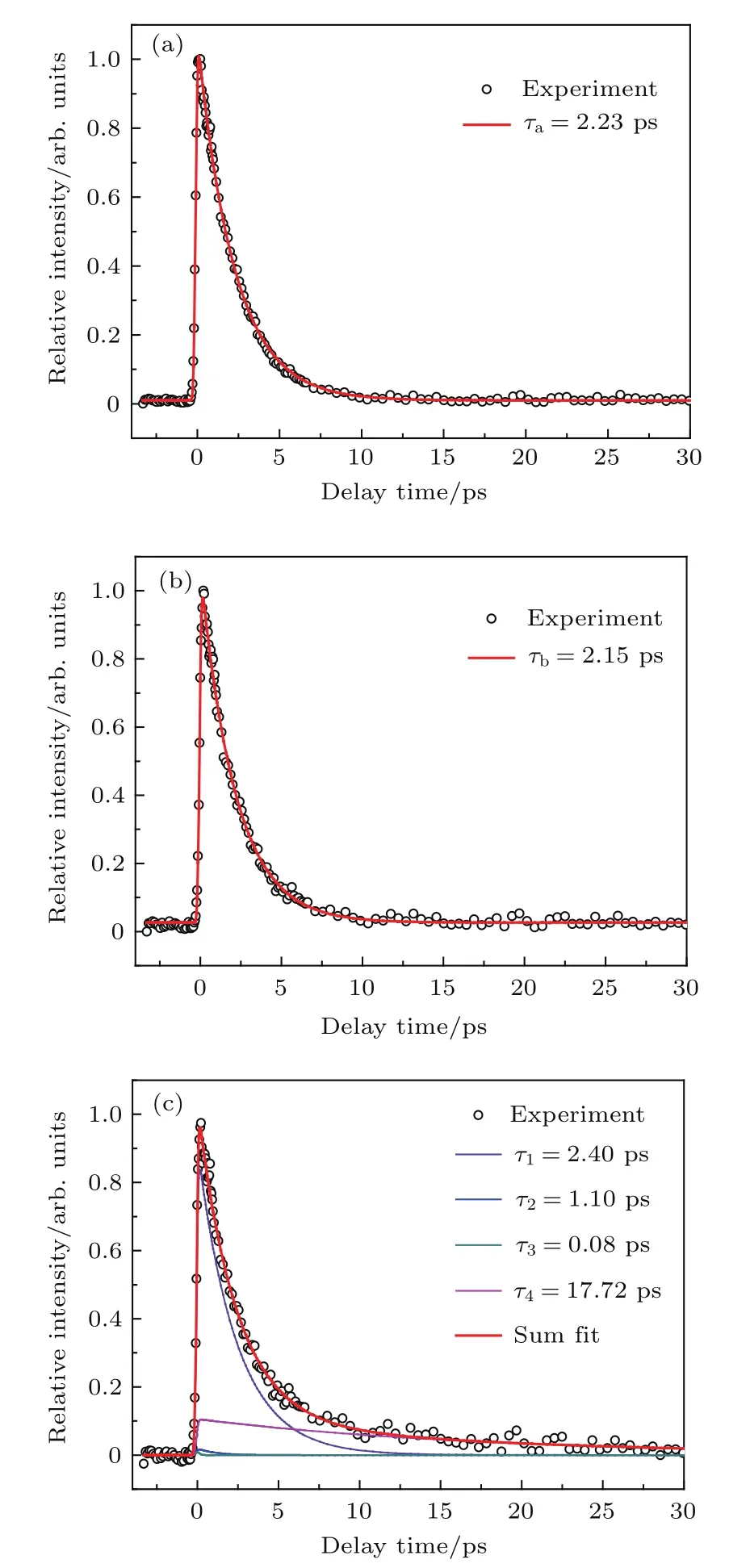
圖3 母體及典型碎片離子的變化曲線 (a)CH3COCH2CH(72 amu);(b)CH3CH2CO+(57 amu);(c)CH3CO+(43 amu).圓圈表示實驗數據,不同顏色的實線代表擬合結果Fig.3.Typical transient obtained by monitoring the mass signal of butanone parent ion and fragment ions:(a)CH3COCH2CH3+(72 amu);(b)CH3CH2CO+(57 amu);(c)CH3CO+(43 amu).Circles represent the experimental data,and the colored lines are the fitting results.
不同于丙酰離子,乙酰離子(圖3(c))的時間線型展現出更為豐富的動力學特征.除了類似于母體離子的τ1組分,還需要再添加其他三個時間組分才能很好地擬合實驗結果.擬合得到四個時間組分分別為τ1=(2.40±0.15)ps,τ2=(1.10±0.25)ps,τ3=(0.08±0.02)ps,τ4=(17.72±0.80)ps,這四個組分的相對比值分別為A1(τ1)=0.46,A2(τ2)=0.01,A3(τ3)=0.01,A4(τ4)=0.05. 由于丁酮對應S2(n,3s)的吸收帶具有明顯的振動結構[13],這說明S2(n,3s)并非直接解離態,而是存在預解離過程.處于S2里德堡態帶源附近的丁酮分子,除了在探測光作用下直接被電離生成母體離子外,也可能通過內轉換弛豫到S1態,并快速地隨機分配S2與S1態之間能量差對應的振動能.處于S1態高振動態的丁酮分子,可能存在電離、解離性電離和兩個α-C-C鍵的解離反應.我們的研究結果顯示(圖3),CH3CO-CH2CH3鍵會發生初次解離生成CH3CO()和CH2CH3,經歷的時間為τ2=(1.10±0.25)ps,代表了處于S1態高振動態上的分子發生初步解離生成激發態的CH3CO()基團所需要的時間.
熱的乙酰基CH3CO()解離過程多年來一直是研究的熱點[22-26].我們根據實驗結果(圖3(c)),分析認為丁酮S1態初步解離的產物CH3CO(),類似于母體離子的S2→S1內轉換過程,通過CCO彎曲振動,迅速內轉換為基態的CH3CO(),所需時間為τ3=(0.08±0.02)ps,也就是說τ3代表了激發態的CH3CO()基團內轉換至基態CH3CO()基團所需要的時間.隨后,基態CH3CO()基團繼續發生二次解離生成CO和CH3,所需要的時間為τ4=(17.72±0.80)ps.丁酮被激發到S2態后的弛豫過程示意圖如圖4所示.丁酮的α-C-C光解離過程受到很多因素的制約,包括探測光的能量、內轉換時間、解離勢壘以及CCO彎曲運動等.如分子的初步解離和→內轉換都是飛秒超快過程,受到緩慢的S2→S1內轉換影響,τ2,τ3具有很大的絕對誤差.
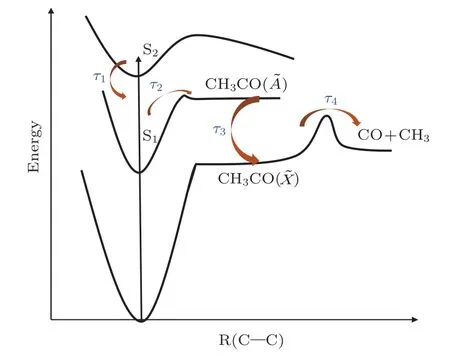
圖4 丁酮分子的弛豫過程示意圖Fig.4.Schematic relaxation processes of butanone.
對于丁酮分子的Norrish I型反應,α-C-C鍵的解離存在分子內振動能量再分配(IVR)與勢壘解離兩種競爭的解離方式[10,11].我們的實驗結果顯示,初步解離和二次解離的時間都達到了皮秒量級,尤其是二次解離的時間高達17.72 ps.對于丁酮分子的初步光解離,通過S2→S1內轉換獲得的大部分振動能量會首先集中于CCO彎曲模式.800 nm的探測光作用于處于S1熱振動態的分子時,1.55 eV的探測光子能量無論對CH3CO-CH2CH3鍵還是CH3-COCH2CH3鍵,都不能使其發生解離,但卻容易被CCO彎曲振動吸收,并進行分子內能振動能量再分配.隨著吸收光子的增加,α-C-C鍵的振動能量在達到3.54 eV時,CH3CO-CH2CH3鍵的解離反應發生.
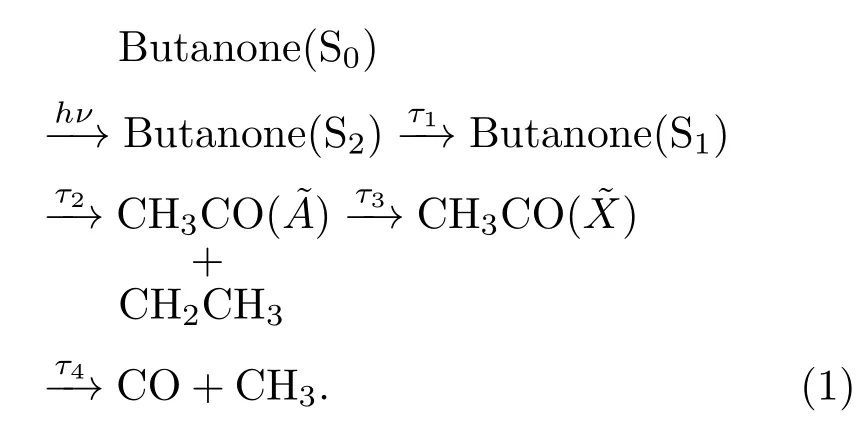
丁酮分子光解離的復雜動力學過程可以通過反應通道(1)所示的動力學模型近似地給出,hν為泵浦光子能量.其中二次解離長達17.72 ps的解離時間很可能是分子內振動能量再分配通道發生解離的結果.而且,分子內振動能量再分配也造成了二次解離時間遠大于初步解離.初步解離時,各振動模式的振動能量較小,分子內振動能量再分配過程顯得緩慢,使得光子能量更容易集中于CCO彎曲振動,解離會在短時間內發生.在二次解離時,由于之前積累了很大的振動能量,分子內振動能量再分配的速度也相應地加快,CH3CO()吸收的探測光子能量容易耗散為其他振動模式,CCO彎曲振動能量達到解離閾值就需要更長的時間.
4 結 論
195.8 nm泵浦光將丁酮分子激發到S2里德堡態后,再吸收3個800 nm探測光子被電離.母體離子的時間線型擬合得到一個約為2.23 ps時間常數,歸屬為S2→S1內轉換的時間.丙酰離子展現出母體離子類似的動力學特征,說明它完全來自于母體離子的解離性電離過程.但乙酰離子的時間線型表現出更豐富的動力學特征,擬合得到四個時間組分,τ1=(2.40±0.15)ps,τ2=(1.10±0.25)ps,τ3=(0.08±0.02)ps,τ4=(17.72±0.80)ps,分別對應S2內轉換至S1,S1發生初步解離生成CH3CO(),CH3CO()內轉換至CH3CO(),CH3CO()發生二次解離生成CO和CH3的時間.其中S2→S1內轉換后,處于S1態高振動態的丁酮分子吸收探測光子后,解離方式上存在分子內振動能量再分配與勢壘解離的競爭.由于較小的探測光子能量(約1.55 eV)容易被CCO彎曲振動吸收,兩個競爭性的解離通道只有CCO彎曲振動導致的解離發生.而且,鍵能最低的CH3CO-CH2CH3鍵具有解離優先權,生成CH3CO()和CH2CH3,所需時間約為1.10 ps.CH3CO()通過CCO彎曲振動,在約0.08 ps的時間內迅速內轉換為CH3CO().基態的CH3CO()與S1態的初步解離類似,通過分子內振動能量再分配通道發生解離,生成CO和CH3,解離時間約為17.72 ps.
感謝中國科學院武漢分院物理與數學研究所波譜與原子分子物理國家重點實驗室的全體同仁在實驗和論文寫作上給予的熱心幫助.
[1]Vacher J R,Jorand F,Blin-Simiand N,Pasquiers S 2008Int.J.Mass Spectrom.273 117
[2]Mu Y,Mellouki A 2000J.Photochem.Photobiol.A134 31
[3]Haas Y 2004Photochem.Photobiol.Sci.3 6
[4]Noyes W A,Porter G B,Jolley J E 1956Chem.Rev.56 49
[5]Diau E W G,K?tting C,ZewailA H 2001ChemPhysChem2 273
[6]Diau E W G,K?tting C,ZewailA H 2001ChemPhysChem2 294
[7]Diau E W G,K?tting C,S?lling T I,Zewail A H 2002ChemPhysChem3 57
[8]S?lling T I,Diau E W G,K?tting C,Feyter S D,Zewail A H 2002ChemPhysChem3 79
[9]Chen W K,Ho J W,Cheng P Y 2003Chem.Phys.Lett.380 411
[10]Chen W K,Ho J W,Cheng P Y 2005J.Phys.Chem.A109 6805
[11]Chen W K,Cheng P Y 2005J.Phys.Chem.A109 6818
[12]Chen W K,Ho J W,Cheng P Y 2005Chem.Phys.Lett.415 291
[13]O Toole L,Brint P,Kosmidis C,Boulakis G,Tsekeris P 1991J.Chem.Soc.,Faraday Trans.87 3343
[14]Loo R O,Hall G E,Houston P L 1989J.Chem.Phys.90 4222
[15]Zou P,McGivern W S,North S W 2000Phys.Chem.Chem.Phys.2 3785
[16]Wei Z,Zhang F,Wang Y,Zhang B 2007Chin.J.Chem.Phys.20 419
[17]Zhang R R,Qin C C,Long J Y,Yang M H,Zhang B 2012Acta Phys.-Chim.Sin.28 522
[18]Sun C K,Hu Z,Yang X,Jin M X,Hu W C,Ding D J 2011Chem.Res.Chinese Universities27 508
[19]Shen L,Zhang B,Suits A G 2010J.Phys.Chem.A114 3114
[20]Traeger J C 1985Org.Mass Spectrom.20 223
[21]Traeger J C,McLouglin R G,Nicholson A J C 1982J.Am.Chem.Soc.104 5318
[22]Owrutsky J C,Baronavski A P 1998J.Chem.Phys.108 6652
[23]Mordaunt D H,Osborn D L,Neumark D M 1998J.Chem.Phys.108 2448
[24]Shibata T,Li H Y,Katayanagi H,Suzuki T 1998J.Phys.Chem.A102 3643
[25]Deshmukh S,Myers J D,Xantheas S S,Hess W P 1994J.Phys.Chem.98 12535
[26]Kroger P M,Riley S J 1977J.Chem.Phys.67 4483
PACS:33.80.Gj,33.20.Lg,82.80.Ms,82.30.Lp DOI:10.7498/aps.66.023301
Ultrafast photodissociation dynamics of butanone in 3s Rydberg state?
Luo Jin-Long1)Ling Feng-Zi2)Li Shuai2)Wang Yan-Mei2)Zhang Bing2)?
1)(Department of Physical Science and Technology,Kunming University,Kunming 650214,China)
2)(State Key Laboratory of Spectroscopyand Atomic and Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,China)
12 September 2016;revised manuscript
1 November 2016)
The initiation and subsequent control or exploration study of chemical transformation in real time by using ultrashort laser pulses aim at femtochemistry.The real-time investigations of ultrafast dynamics of excited molecules in gas and condensed phases have attracted a great deal of attention over the last two decades.As a kind of important organic compound,aliphatic ketone is an area of much interest for many research fields,especially for atmospheric photochemistry.Via photodissociation reaction,it can release carbonyl radical whose chemical character is active and can react with hydroxyl easily.As a typical aliphatic ketone,butanone has been a research focus over the past decades.The ultrafast dissociation dynamics of butanone after excitation to the second electronically excited state(S2)with a 195.8 nm pump pulse is studied by the femtosecond pump-probe technique combined with the time-of-flight mass spectrometry(TOF-MS).Time-resolved mass spectrometry(TRMS)has proven to be a powerful technique to study the ultrafast dynamics of excited states in molecules.In this technique,the MCP detector is capable of recording time-resolved ion yield measurements of different cations by monitoring the current output directly from the anode by using an oscilloscope.This enables a time-of-flight mass spectrum to be recorded at each delay time,which is controlled by a delay stage,and the measured total signal is then integrated,yielding a time-resolved ion yield transient,which is conducted by LABVIEW software.The pump wavelength in this work is set to be 195.8 nm and the probe laser wavelength is centered at 800 nm.The complex ultrafast dynamics in butanone with 3s Rydberg state excitation and its possible decay paths and following dissociation mechanism are given.Experimental results show that the Norrish I type dissociation kinetics of butanone exhibit rich features,for it has a methyl group and an ethyl group atαposition.The decay time constant of the parent transient is approximately 2.23 ps±0.02 ps.There is only one time constant of 2.15 ps±0.02 ps for the fitting of the propionyl transient.The best fit of acetyltransient is obtained with four time constants:τ1=(2.40±0.15)ps,τ2=(1.10±0.25)ps,τ3=(0.08±0.02)ps,andτ4=(17.72±0.80)ps,corresponding to S2→S1internal conversion,the primary dissociation of the S1state generating CH3CO(),→internal conversion and secondary dissociation of CH3CO()respectively.Two competitiveα-CC bond dissociation processes are observed and discussed.They are dissociation channels through intramolecular vibrational energy redistribution(IVR)and/or by getting over the dissociation barrier inα-cleavage of butanone.But hereunder the condition of this experiment,the dissociation is the result of IVR.
time-resolved mass spectrum,ultrafast dynamics,pump-probe,photodissociation
:33.80.Gj,33.20.Lg,82.80.Ms,82.30.Lp
10.7498/aps.66.023301
?國家重點基礎研究發展計劃(批準號:2013CB922200)和國家自然科學基金(批準號:21573279,21273274)資助的課題.
?通信作者.E-mail:bzhang@wipm.ac.cn
*Project supported by the National Basic Research Program of China(Grant No.2013CB922200)and the National Natural Science Foundation of China(Grant Nos.21573279,21273274).
?Corresponding author.E-mail:bzhang@wipm.ac.cn
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
艦船科學技術(2022年8期)2022-06-05 07:36:28
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
瘋狂英語·新讀寫(2020年3期)2020-06-06 09:05:56
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
中國公路(2017年18期)2018-01-23 03:00:38
數學物理學報(2017年6期)2018-01-22 02:26:40
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
