磷脂微乳電動色譜的定量結構保留關系研究
2017-06-21 07:51:38宋靜鄭園霍長虹劉建芳
分析化學 2017年5期
宋靜+鄭園+霍長虹+劉建芳

摘 要 以大豆磷脂為主要的表面活性劑,制備適合毛細管電動色譜使用的不同構成比的微乳體系, 應用溶劑化參數模型研究了中性溶質在其中的定量結構保留關系。使用動態涂層毛細管, 以二甲基亞砜和十二烷基苯分別作為電滲流和微乳液滴遷移的標記物, 測定了26個具有不同結構小分子中性化合物在17種微乳電動色譜體系下的保留因子, 建立了線性溶劑化能量關系(LSER)方程。通過比較兩體系的LSER方程系數比較體系相似性。結果表明, 本研究建立的磷脂微乳電動色譜體系在線性溶劑化特征上和其它構成的微乳電動色譜體系相似。對溶質保留貢獻較大的是溶質體積和有效氫鍵堿度, 油相種類及濃度對溶質的保留選擇性無明顯影響。
關鍵詞 微乳電動色譜; 大豆磷脂; 保留因子; 線性溶劑化能量關系
1 引 言
微乳電動色譜(Microemulsion electrokinetic chromatography, MEEKC)是使用微乳作為分離介質的電泳技術, 具有分離效率高、適用范圍廣和樣品消耗少等優勢, 已廣泛應用于分離科學領域[1,2]。中性物質在MEEKC系統中依據其在水相和假固定相(微乳液滴)的分配系數不同而實現分離, 而帶電物質除了在兩相間的分配系數不同外, 還有物質間的靜電作用。MEEKC的選擇性通過假固定相的改變實現, 不同的表面活性劑、油相、助表面活性劑和有機改性劑的加入都會改變微乳對分離物的選擇性。盡管MEEKC的應用已有20余年, 已有許多關于微乳體系構成的報道[3], 但最常用的仍是以十二烷基硫酸鈉(Sodium dodecyl sulfate, SDS)作為表面活性劑制備的微乳。Lucangioli等[4,5] 最初將磷脂類生物表面活性劑引入MEEKC體系中, 并將其應用于免疫抑制劑倍他米松及其衍生物的正辛醇/水分配系數(nOctanol/water partition coefficient, lgP)測定, 發現用磷脂類生物表面活性劑制備的微乳體系預測以上藥物的lgP值更準確。但是該研究僅檢測了4種藥物, 且未見應用該系統進行其它藥物集合lgP值測定的報道, 因此磷脂類微乳電動色譜體系提高藥物lgP值預測準確性的結論尚不十分肯定。磷脂類微乳與其它構成的微乳體系是否存在保留機制的差別也未見報道。
線性溶劑化能量關系(Linear solvation energy relationship, LSER)模型廣泛用于各種色譜體系的溶質保留機制研究[6~8]以及正辛醇水分配系數[7]和藥物體內透膜過程的預測[9,10]。通過方程系數間的比較, 可以發現不同分配體系的相似性, 選擇合適的色譜體系用于藥物膜通透性的篩選[7,11]。本研究采用LSER模型, 對應用大豆磷脂制備的微乳體系進行MEEKC分離時的溶質保留機制進行研究, 比較了不同磷脂構成比、不同油相及油相含量對LSER方程系數的影響, 同時將磷脂微乳體系與其它表面活性劑構成的微乳或膠束體系進行了比較, 擬闡明磷脂類成分和油相組分在微乳分離體系中的作用, 為進一步應用磷脂類微乳電動色譜體系進行藥物膜通透性的預測提供理論依據。
2 實驗部分
2.1 儀器與試劑
Beckman P/ACE MDQ毛細管電泳儀、DAD檢測器(美國Beckman公司);彈性石英毛細管柱(河北永年光導纖維廠, 60 cm×50 μm, (id), 有效長度50 cm);Malvern Zetasizer Nano ZS90型激光粒度散射儀(英國Malvern公司);KQ3200B型超聲波清洗器(昆山市超聲儀器有限公司);BP211D電子分析天平(德國賽多利斯公司); pH3C型pH計(上海雷磁儀器廠)
十二烷基硫酸鈉(石家莊市拜昂生物技術有限公司);聚凝胺(PB, 美國Sigma公司);葡聚糖硫酸鹽(DS, 美國Sigma公司);大豆磷脂(注射級, 上海太偉藥業股份有限公司);膽酸鈉(美國Sigma公司, BioXtra ≥ 99%);脫氧膽酸鈉(SigmaAldrich 公司);十七氟辛烷磺酸四乙基銨鹽(Aldrich 公司);十六烷三甲基溴化銨、十二烷基苯(Sigma公司);肉豆蔻酸異丙酯(上海源葉生物科技有限公司);正丁醇、正辛醇、正庚烷(分析純, 天津市永大化學試劑有限公司);全氟辛基磺酸鉀(Aldrich 公司);二甲基亞砜(DMSO, 分析純, 天津市標準科技有限公司);甲醇(色譜純, 天津市康科德科技有限公司);實驗用水為某品牌純凈水。
待測化合物見表1, 其中間二甲苯、對二甲苯由河北科技大學提供, 其余均由河北醫科大學藥學院提供。
2.2 微乳和化合物儲備液的制備
優化的微乳處方構成見表2。稱取適量的表面活性劑于密封性容器內, 用水相(20 mmol/L NaH2PO4)溶解, 依次加入助表面活性劑、油相, 混勻、超聲30 min, 室溫靜置(12 h 以上)至微乳溶液搖晃后仍澄清, 表明形成穩定微乳, 最后用飽和NaOH調至所需pH值, 備用, 使用前用0.45 μm濾膜過濾。
根據Vitha等[12]對建立溶劑化方程的溶質集合篩選提出的指導原則, 共收集了26個具有不同結構特征的小分子中性化合物及其Abraham分子描述符(見表1), 分別用甲醇溶解, 配制成3~100 mg/mL儲備液, 進樣前用微乳液稀釋至0.1~3 mg/mL。
1 mL微乳液中加入6 μL苯甲酰胺甲醇溶液(30 mg/mL)、6 μL鄰甲苯胺甲醇溶液(100 mg/mL)、4 μL 2萘酚甲醇溶液(100 mg/mL)和6 μL 電滲流標記物DMSO、4 μL 微乳標記物十二烷基苯, 作為標準混合液。
2.3 保留因子的測定
2.3.1 毛細管預處理 (1)毛細管活化 用甲醇沖洗新管3 min, 再依次用水沖洗0.5min、 0.1 mol/L HCl 沖洗15 min、水沖洗0.5 min、1 mol/L NaOH 沖洗15 min。(2)毛細管涂層 毛細管活化后靜置30 min, 先用5% PB溶液沖洗20 min, 靜置20 min后, 再用3%DS溶液沖洗20 min, 靜置30 min, 使用前用水沖洗5 min即可。
2.3.2 測定條件 毛細管柱總長度60 cm(有效長度50 cm, 內徑50 μm), ±20 kV恒壓分離, 檢測波長208 nm, 壓力進樣(0.8 psi, 8 s), 柱溫25℃。
2.3.3 不同MEEKC體系下保留因子的測定與計算 陰離子型微乳(表2中ME1ME16)采用+20 kV電壓, 陽離子型微乳(表2中ME17)采用20 kV電壓分離, 柱溫25℃。保留因子k按下式計算 [13]:
2.3.4 保留因子與Abraham描述符間LSER的建立 LSER理論是基于腔穴模型, 解釋溶質在兩相間轉移[14], LSER方程一般描述如下:
SP作為給定系統的一種溶劑化性能, 例如溶劑的保留行為的對數(lgk)、正辛醇/水分配系數(lgP)、穩態血腦藥物濃度比(lgB)等;方程中的大寫字母是Abraham溶質描述符, E表示摩爾折射率, S表示溶質偶極/極化率, B和A分別表示有效氫鍵堿度和酸度, V代表特征體積;小寫字母 e, s, a, b和v是通過多元線性回歸得到的各個變量的系數, 反映了給定系統的性質即溶劑化參數對系統的貢獻大小, 其中e代表分配系統中的溶質分子與溶劑的π或n電子的作用;s反映系統兩相間的偶極/極化率;b和a分別代表系統的有效氫鍵堿度和酸度;v代表系統的空穴的形成能力或內聚能;常數c是方程的截距, 由溶質和系統間恒定的相互作用產生, 如相體積比。
本研究將不同微乳體系下測得的26個化合物的保留因子(lgk)與Abraham描述符間建立了LSER方程。
3 結果與討論
3.1 微乳體系構成
通過改變表面活性劑及油相的種類和濃度制備的穩定微乳處方組成見表 2。
文獻[15]報道, 制備微乳時各組分的加入順序會影響微乳的形成, 但是本實驗發現, 超聲和靜置有助于微乳的形成, 即使改變組分的加入順序, 仍能夠形成穩定的微乳體系。微乳制備和保存都應在密封的容器內進行, 以防止油相和丁醇等成分的揮發影響微乳的構成。運行MEEKC時, 洗脫時間窗口的大小與微乳中助表面活性劑及表面活性劑的濃度相關, 本研究控制時間窗都在20 min以上, 以保證洗脫窗內能夠容納較多的溶質。
3.2 微乳穩定性
通過外觀、粒徑和電位變化考察了微乳的穩定性, 部分檢測結果見表3。從表中粒徑和Zeta電位值可知, 微乳室溫存放30天, 粒徑和表面電位無明顯變化。
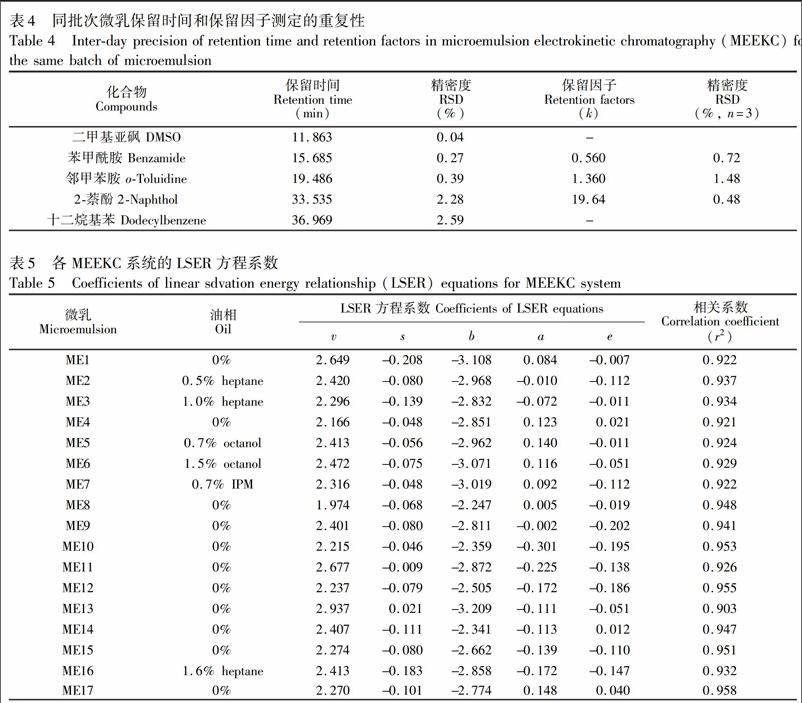
3.3 溶質保留時間和保留因子重現性
以ME5為電動色譜流動相, 用標準混合液進樣, 考察動態涂層條件下溶質保留時間的重復性, 圖1是典型的色譜圖。結果表明, 應用文獻[16]報道的涂層方法(見2.3.1節毛細管涂層), 使用同批次微乳液, 連續測定3天, 總進樣60針(每針運行時間在20 min以上), 化合物的遷移時間RSD<2.5%。隨著進樣針數的增加和進樣時間的延長, 遷移時間RSD變大, 原因可能是樣品在毛細管內壁的吸附或涂層有損壞。因此測定樣品過程中, 需要用標準混合溶液進行重復性檢測, 如標準樣品的遷移時間出現較大偏差, 則需對毛細管進行沖洗;沖洗后無改善則需重新涂層處理, 以確保溶質遷移時間的重復性。從表4可見, 各被測物保留時間與保留因子測定的重復性均良好。但為了保證保留因子測定的準確性, 每個樣品溶液中都加入DMSO和十二烷基苯, 以盡量減少遷移時間變化帶來的保留因子測定誤差。
3.4 不同微乳體系下的LSER方程
收集26個不同結構的小分子中性化合物, 以表2中的微乳體系作為分離介質, 測定溶質的保留因子, 用SPSS 19.0進行保留因子與溶質描述符間的多元線性回歸, 建立LSER方程, 結果見表5。
總體上看, 實驗中得出的各個微乳電動色譜體系的LSER方程系數都比較接近, 其中v和b的系數絕對值較大, 表明溶質的體積和氫鍵堿性對溶質保留貢獻較大。 v絕對值較大表明體系中油滴形成腔穴對溶質保留所需能量低于水相; b值為負值表明微乳氫鍵堿性小于緩沖液, 隨著溶質氫鍵堿性的增加將不利于溶質進入假固定相中。 a值在各體系LSER方程中大部分為負值且接近于0, 表明溶劑氫鍵受體能力在油水兩相間的性能相似; s和 e的絕對值較小, 表明在溶質保留機制中偶極/極化率和溶質分子與溶劑的π或n電子的作用較弱[7,17]。
微乳相對于膠束溶液來說, 主要不同在于加入了助表面活性劑和油相[18], 本研究系統比較了不同油相種類和含量對體系特征的影響。從表5中ME1~ME3, ME4~ME6體系的LSER方程可見, 在其它組成不變的情況下, 庚烷從0到1.0%, 辛醇從0到1.5%, LSER系數沒有明顯變化。 從ME2, ME5, ME7的方程系數可見, 不同油相種類(庚烷、辛醇、IPM)對體系特征也無影響。這些結果表明油相對MEEKC測定中性化合物的分離選擇性沒有明顯影響。若待測化合物有一定的解離性, 則其分離選擇性可能與油相有關[2]。助表面活性劑一般為短鏈醇, 它的加入可以改變微乳液滴的表面電荷密度和微乳結構的剛性, 有利于溶質的溶解和洗脫[10]。
表面活性劑的種類直接影響微乳的表面特性。SDS是MEEKC系統常用的表面活性劑, 本實驗對以大豆磷脂和碳氟表面活性劑作為表面活性劑的新型微乳體系的溶劑化特征進行了研究(ME1, ME4, ME8, ME17)。結果表明, 大豆磷脂的加入, 沒有改變微乳體系的基本特征。而據文獻[19,20]報道, 在碳氟表面活性劑構成的膠束溶液中, 因碳原子上的所有氫原子被氟原子取代, 在溶質分配行為中氫鍵酸度作用貢獻增加, 甚至強于氫鍵堿度, 與SDS膠束體系中氫鍵酸度貢獻很小的結果顯著不同。但本研究未得到一致結果, 在PFOSK或PFOSNH4形成的微乳體系中, 依然是氫鍵堿度的作用突出。這可能是因為本研究PFOSK或PFOSNH4體系中加入了SDS、SDC和丁醇等, 而SDS和SDC的氫鍵堿度作用貢獻較大。
此外, 本實驗還發現, 陽離子型表面活性劑CTAB建立的MEEKC體系的LSER方程系數與陰離子型表面活性劑構成MEEKC體系(ME1ME9)相比也無較大差異, 表明微乳液滴只是作為一個相對于水相極性較弱的假固定相, 其表面電荷性質不影響中性化合物的分離選擇性。
3.5 與文獻報道的其它色譜系統的比較
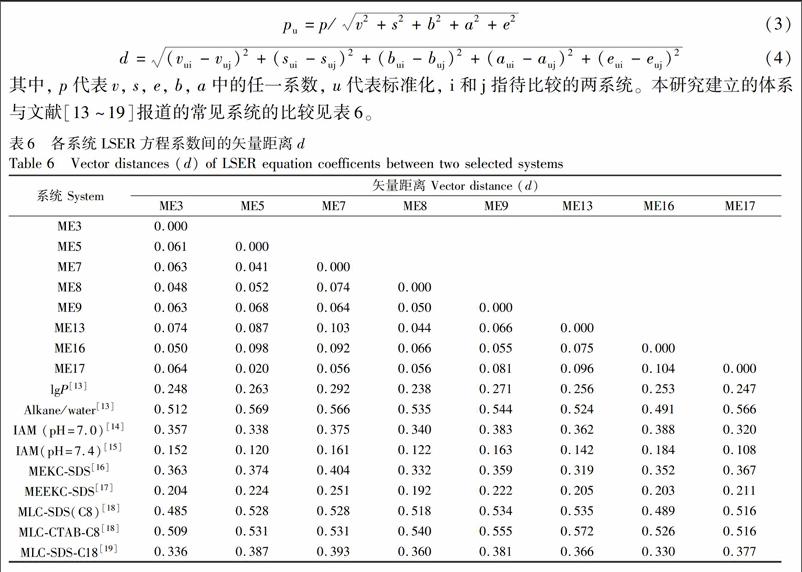
其它應用LSER模型研究溶質保留機制的色譜系統有磷脂膜色譜、膠束、微乳電動色譜系統等[21~27], 本研究應用Lázaro 等提出的矢量距離法[28]比較了所建新型MEEKC系統與其它色譜系統間的接近程度。距離參數d的計算方法如下方程:
其中, p代表v, s, e, b, a中的任一系數, u代表標準化, i和j指待比較的兩系統。本研究建立的體系與文獻[13~19]報道的常見系統的比較見表6。
根據文獻[6,29]報道, 系統間的距離d<0.25, 表明兩系統特征較相似。從表6可見, 所建立的各種微乳體系彼此間距離d<0.1, 說明彼此間特征相近。所建微乳體系與正辛醇/水分配系數(lgP)、MEEKCSDS和磷脂膜色譜(pH 7.4)系統間的d值均約為0.25, 表明MEEKC、磷脂膜色譜與正辛醇水系統所測得的親脂性參數一致, 表明MEEKC是預測lgP值的實用工具[7]。本研究中所用的磷脂微乳體系與其它MEEKC體系相比, 基線穩定性更好, 分離能力更強。
4 結 論
對于本研究測定的中性化合物, 無論是在大豆磷脂、SDS、CTAB, 還是碳氟類表面活性劑制備的微乳電動色譜中, 所得保留因子的LSER方程的基本特征相似, 都是v和b較大, 即溶質的體積和氫鍵堿度對其在微乳中的保留影響較大, 前者利于保留, 后者減弱保留。各體系之間距離也非常接近, 說明表面活性劑類型和油相組成對中性溶質在微乳體系中的保留選擇性均無明顯影響。若不考慮油相對微乳液滴剛性結構的影響, 微乳體系或許可以稱為助表面活性劑修飾的膠束體系。
References
1 Chang C W, Chen Y C, Liu C Y. Electrophoresis, 2015, 36(21): 2745-2753
2 Xiao W, Zhang Q, Chen C, Zhang Q H, Hu Y J, Xia Z N, Yang F Q. J. Pharm. Sci, 2016, 54(9): 1678-1686
3 Buchberger W. Method Mol. Biol., 2016, 1483: 91-109
4 Lucangioli S E, Kenndler E, Carlucci A, Tripodi V P, Scioscia S L, Carducci C N. J. Pharm. Biomed. Anal., 2003, 33(5): 871-878
5 Lucangioli S E, Carducci C N, Scioscia S L, Carlucci A, Bregni C, Kenndler E. Electrophoresis, 2003, 24(6): 984-991
6 Li L X, Yang J R, Huang H Z, Xu L Y, Gao C K, Li N. Biomed. Chromatogr., 2016, 30(7): 996-1006
7 Subirats X, Yuan H P, Chaves V, Marzal N, Rosés M. Electrophoresis, 2016, 37(14): 2010-2016
8 Janicka M. J. Chromatogr. Sci., 2014, 52(7): 676-684
9 Baynes R E, Xia X R, Vijay V, Vijay J E. SAR QSAR Environ. Res., 2008, 19(78): 615-630
10 Stepnik K E, Malinowska I. J. Chromatogr. A, 2013, 1286: 127-136
11 Liu J F, Sun J, Sui X F, Wang Y J, Hou Y N, He Z G. J. Chromatogr. A, 2008, 11981199: 164-172
12 Vitha M, Carr P W. J. Chromatogr. A, 2006 1126(12): 143-194
13 Jiang Z J, Reilly J, Everatt B, Briard E. J. Pharm. Biomed. Anal., 2011, 54(4): 722-729
14 Lesellier. E. J. Chromatogr. A, 2015, 1389: 49-64
15 Liu J F, Sun J, Wang Y J, Li H Y, Sui X F, Li Y, He Z G. Asian J. Pharmaceut. Sci., 2008, 3(4): 158-167
16 Katayama H, Ishihama Y, Asakawa N. Anal. Chem., 1998, 70(11): 2254-2260
17 LI Jie, SUN Jin, HE ZhongGui. Acta Pharmaceutica Sinica, 2007, 42(1): 13-18
李 潔, 孫 進, 何仲貴. 藥學學報, 2007, 42(1): 13-18
18 LIU JianFang, SUI XiaoFan, SUN Jin, WANG Yongjun, HE ZhongGui. J. Anal. Chem., 2008, 36(12): 1742-1748
劉建芳, 隋曉璠, 孫 進, 王永軍, 何仲貴. 分析化學, 2008, 36(12): 1742-1748
19 Esaka Y, Rin F, Kobayashi M, Osako R, Murakami H, Uno B. J. Chromatogr. A, 2014, 1385(2): 261-268
20 Fuguet E, Ràfols C, Bosch E, Rosés M, Abraham M H. J. Chromatogr. A, 2001, 907(12): 257-265
21 Abraham M H, Chadha H S, Whiting G S,Mitchell R C. J. Pharm. Sci., 1994, 83(8): 1085-1100
22 Li J, Sun J, Cui S M, He Z G. J. Chromatogr. A, 2006, 1132(12): 174-182
23 Valko K, Du C M, Bevan C D, Reynolds D P, Abraham M H. J. Pharm. Sci., 2000, 89(8): 1085-1096
24 Trone M D, Khaledi M G. Anal. Chem., 1999, 71(7): 1270-1277
25 Abraham M H, Treiner C, Rosés M,Rafols C, Ishihama Y. J. Chromatogr. A, 1996, 752(12): 243-249
26 García M A, Vitha M F, Sandquist J, Mulville K, Marina M L. J. Chromatogr. A, 2001, 918(1): 1-11
27 García M A, Vitha M F, Marina M L. J. Liq. Chrom. Rel. Technol., 2000, 23(6): 873-895
28 Lázaro E, Ràfols C, Abraham M H, Rosés M. J. Med. Chem., 2006, 49(16): 4861-4870
29 Poole S K, Poole C F. J. Chromatogr. A, 2008, 1182(1): 1-24
