降脂紅曲產品中Monacolin K檢測方法的比較
2017-03-21 10:19:12趙光隆張薄博許贛榮
食品與生物技術學報 2017年1期
趙光隆, 張薄博, 許贛榮
降脂紅曲產品中Monacolin K檢測方法的比較
趙光隆, 張薄博, 許贛榮*
(江南大學 生物工程學院,江蘇 無錫 214122)
為準確測定降脂紅曲類產品中Monacolin K(MK)的質量濃度。作者對中華人民共和國輕工行業標準(2007)、保健食品檢驗與評價技術規范(2003版)、臺灣食品中MK檢驗方法(2012)以及其它方法進行比較。確定朱華等的檢測方法較優,MK在質量濃度10~400 μg/mL范圍內線性良好,加標回收率為96.3%,方法精密度RSD=1.6%。該方法簡單、快捷,能夠比較準確地反映紅曲類產品中不同構型MK質量濃度,具有很高的實用價值。
降脂紅曲;莫納可林K;檢測方法
降脂紅曲是經過紅曲菌發酵,富含降血脂活性物質Monacolin K(以下簡稱MK)的紅曲產品。自從1979年,日本遠藤章[1]從紅色紅曲菌(Monascus ruber)的培養液中獲得一種膽固醇合成抑制劑,并命名為Monacolin K(中文名,莫納可林K),后被證實與降血脂藥物洛伐他汀 (lovastatin)為同一種物質。它能有效抑制膽固醇合成過程中關鍵酶HMGCoA(3-羥基-3-甲基-戊二酰基-輔酶A還原酶)的活性[2]。
MK有兩種分子構型:C24H38O6(酸式構型)、C24H36O5(內酯式構型),結構式見圖1。微生物發酵法得到的都是酸式結構的MK,它可以直接溶于體液并發揮生理作用;但它比內酯式的穩定性差,內酯式的MK要在體內先轉化成酸式的才能被吸收并發揮作用[3]。

圖1 MK分子結構Fig.1 MK molecular structure
紅曲菌和土曲霉都可發酵產生降脂活性成分MK。微生物發酵法合成的MK是酸式結構。但為了保證洛伐他汀的穩定性,在土曲霉生產洛伐他汀的提取純化過程中將其轉化成內酯式結構。降脂紅曲產品一般是通過固態發酵來生產的,無需復雜的后處理就可直接服用,故產品中保留了大量的酸式MK,但在生產過程中,由于烘干貯存等過程,酸式MK會自然轉變成內酯式結構[4]。一般情況下固態發酵的降脂紅曲產品中酸式MK和內酯式MK共存。
目前國內市場上有多種類型的降脂紅曲產品:中成藥、中藥飲片、保健食品。對這些產品中MK的測定,也是依據各自的標準采用不同的方法來檢測。這些標準分別是:中華人民共和國輕工行業標準(2007)[5](以下簡稱輕工方法)、保健食品檢驗與評價技術規范 (2003版)[6](以下簡稱保健食品方法)、臺灣食品中MK之檢驗方法(2012)[7](以下簡稱臺灣方法)。此外朱華等在2005年報道過的一種方法[8](以下簡稱朱華等的方法)。
從檢測技術的角度來看,仔細考察上述4種檢測MK的方法,發現有相同之處,也有不同之處。保健食品方法中MK的檢測方法參考美國藥典[9]中洛伐他丁的檢測方法,只測定內酯式MK。即使降脂紅曲產品中含有酸式結構的MK,用此法也無法得出酸式結構MK的含量。還有一個值得注意的問題是,不同的方法采用的萃取溶劑種類不同,或者樣品萃取時間有長有短。顯然,依據不同的標準,就有不同的方法檢測MK,這就很可能出現一個問題,即按照不同的方法檢測同一個產品,可能會得到不同的檢測數據。
因此,作者對同一批降脂紅曲產品,按照上述4種不同的方法進行了測定,根據檢測數據對不同的檢測方法展開討論和比較,從而為建立更為科學、規范的降脂紅曲活性成分MK的檢測方法奠定基礎。
1 材料與方法
1.1 儀器與設備
Waters液相色譜儀:色譜柱:Eclipse XDB-C18(4.6 mm×250 mm;5 μm);Waters公司產品。
1.2 試劑
洛伐他丁標準品:中國食品藥品檢定研究院提供。
2 檢測方法
2.1 標準溶液制備
取MK內酯型標準品20 mg,精確稱量,以乙腈溶解并定容至10 mL,作為標準原液,于-20℃避光保存。
2.1.1 MK內酯型標準溶液制備 取一定量標準原液以乙腈稀釋至5.0~400.0 μg/mL,MK內酯型標準溶液。
2.1.2 MK酸型標準溶液 取標準原液1 mL于0.1 mol/L氫氧化鈉溶液1 mL,旋渦混合均均,于50℃超音波水浴振蕩反應1 h,冷卻至室溫后,以乙腈定容至10 mL,繼以乙腈稀釋至5~418 μg/mL,供作MK酸型標準溶液。內酯式MK相對分子質量為404.55,酸式MK相對分子質量為422.57,故經過換算得此時的酸式MK的濃度。y為檢測峰面積,x為對應MK質量濃度,繪制標準曲線。
酸式:y=74 175x-77 348,R2=0.999 8,準確檢測下限10.4 μg/mL
內酯式:y=75 873-87 703,R2=0.999 1,準確檢測下限10.0 μg/mL
從原理上推測,用內酯式公式足以代表酸式的計算。
2.2 各標準中的檢測方法
2.2.1 中華人民共和國輕工行業標準(2007)中的檢測方法 提取條件:將紅曲米粉碎(40目,粉狀)并充分混合均勻。準確稱取400.0~600.0 mg試樣于50 mL容量瓶中,加入30 mL 75%乙醇(體積分數),搖勻,室溫下超聲50 min。加75%乙醇至接近刻度,再超聲10 min,之后冷卻至室溫,用75%乙醇定容至50 mL。以3 500 r/min的旋轉速度離心10 min。取上清液經0.45 μm過濾,濾液待用[5]。
色譜條件:色譜柱:EclipseXDB-C18(5 μm,4.6 mm×250mm);流動相:V(甲醇)∶V(水)∶V(磷酸)= 385∶115∶0.14(體積分數);流量:1.0 mL/min;紫外檢測波長238 nm;進樣量:20 uL;柱溫:25℃[5]。
2.2.2 保健食品檢驗與評價技術規范 (2003版)的檢測方法 提取條件:將片劑、膠囊或紅曲發酵產物試樣粉碎并混合均勻,根據樣品中洛伐他汀含量準確稱取一定量試樣于50 mL試管中,加入10.0 mL pH=3磷酸水溶液。超聲提取10 min后再加人10.0 mL三氯甲烷,置于渦旋混勻器3 min。靜置后去掉上層水相,將三氯甲烷層以3 000 r/min離心3 min。準確吸取上清液1.0~5.0 mL試管中,將試管置于50℃左右水浴中使用真空泵減壓干燥至揮去全部溶劑。向試管中加入流動相并定容至5.0 mL,徹底混勻,經0.45 μm濾膜過濾后待進樣[6]。色譜條件:同輕工方法。
2.2.3 臺灣MK之檢測方法(2012) 提取條件:檢體均質混勻后稱取0.2 g,精確稱定,置于25 mL定量瓶中,加入甲醇約25 mL,旋渦混合均勻后,于室溫超聲波水浴中震蕩萃取30 min,冷卻至室溫后,以甲醇定容。以3 000 r/m離心10 min,取上層液,以濾膜過濾,供作檢液[7]。
色譜條件:色譜柱:EclipseXDB-C18(5 μm,4.6 mm×250 mm);流動相:V(乙腈)∶V(0.1%磷酸溶液)=65∶35;流量:1.5 mL/min;紫外檢測波長238 nm;進樣量:10 μL;柱溫:25℃[7]。
2.2.4 朱華等的檢測方法 提取條件:乙醇提取法:精確稱取0.5 g樣品于50 mL標準比色管中,用體積分數70%乙醇定容至50 mL,55℃水浴浸提1 h,其間每隔20 min震蕩搖勻一次。浸提結束取出冷卻至室溫,取上清液微濾待測[8]。
色譜條件:色譜柱:EclipseXDB-C18(5 μm,4.6 mm×250 mm);流動相:V(乙腈)∶V(0.1%磷酸溶液)=55∶45;流量:1 mL/min;紫外檢測波長238 nm;進樣量:20 μL;柱溫:30℃[8]。
3 結果與討論
3.1 內酯式MK加標回收率實驗
為了探究4種方法中不同的提取條件對樣品提取效果如何,作者進行了加標回收實驗,稱取同一批紅曲粉3份,分別加入3個梯度的內酯式MK,進行加樣內酯式MK回收實驗,統一采用本實驗色譜條件檢測,結果如表1所示,4種標準的檢測方法中,保健食品方法回收率最低且相對標準偏差較大無法滿足檢測準確性的要求。臺灣方法中提取時間較短,回收率偏低。輕工方法與朱華等的方法樣品提取回收率都比較高,回收率≥95.0%,均滿足樣品檢測準確性要求,樣品檢測穩定性朱華等的方法RSD=1.6%優于輕工行業標準
3.2 流動相的比較實驗
流動相作為液相檢測中非常重要的的因素,不同的流動相成分以及不同成分的比例對樣品的出峰時間和峰形有直接影響,直接影響到樣品檢測的準確性。實驗對不同檢測方法中的流動相對標準品的分離效果較好進行比較。輕工方法和保健食品方法:V(甲醇)∶V(水)∶V(磷酸)=385∶115∶0.14流動相,從標準品圖譜(圖2)來看,酸式MK Rt=11.638,內酯式MK Rt=13.296,色譜峰相鄰,從混合標樣來看,兩峰剛好分開,但是在實際樣品檢測中,由于樣品成分復雜和外界條件干擾等因素峰形會發生前沿或拖尾現象,使得兩峰無法完全分開。臺灣方法:V(乙腈)∶V(0.1%磷酸溶液)=65∶35,標準樣品圖譜 (圖3),酸式MK Rt=4.345,內酯式MK Rt=7.605,流動相中乙腈含量過高導致MK的保留時間比較短,在實際樣品檢測中會與樣品中的雜質和溶劑峰混雜在一起,影響含量的準確測定。朱華等的方法:V(乙腈)∶V(0.1%磷酸溶液)=55∶45,從圖譜來看(圖4),降低流動相中乙腈的含量,延長了MK的出峰時間,有效的避免了雜質和溶劑峰的干擾且峰形良好,分離效果較好。
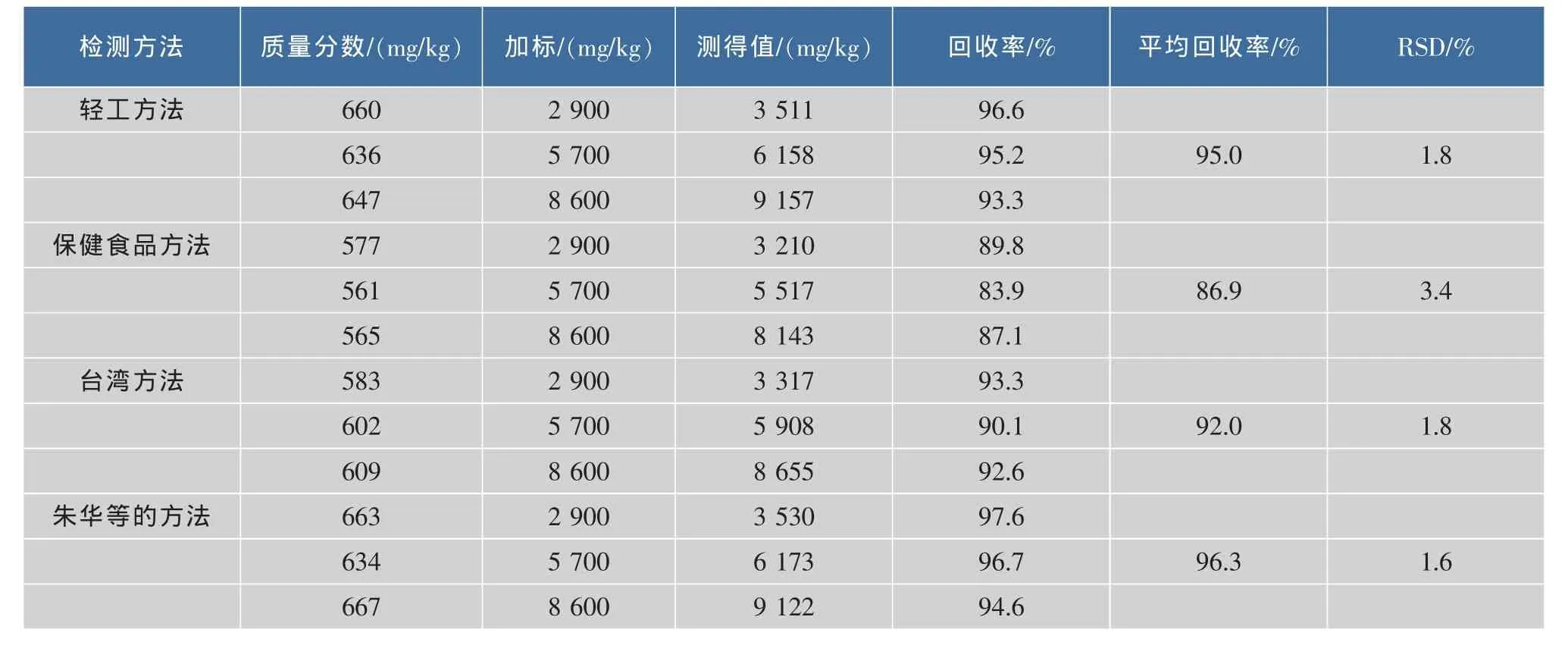
表1 內酯式MK回收率實驗比較Table 1 Compares the lactone type MK recovery experiment
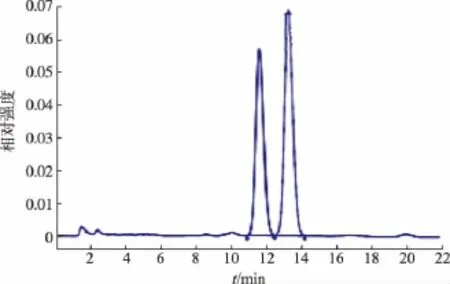
圖2 輕工和保健食品方法混合標樣圖譜Fig.2 Light Industry and Health food industry method HPLC analysis
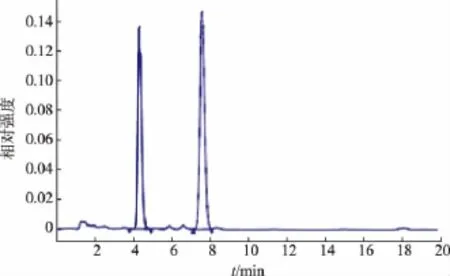
圖3 臺灣方法混合標樣圖譜Fig.3 Taiwan method HPLC analysis
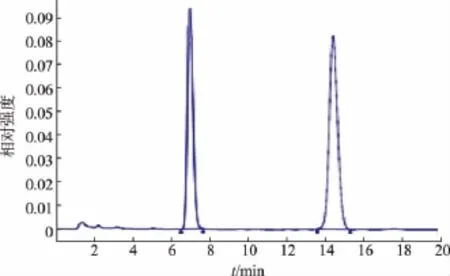
圖4 朱華等方法混合標樣圖譜Fig.4 Zhu Hua et al.applied method HPLC analysis
3.3 樣品檢測實驗
稱取同一批降脂紅曲粉3份,分別用4種標準中的方法進行檢測,結果表明:輕工方法紅曲粉中MK同時以酸式和內酯式的2種形式存在,提取相對比較徹底,液相檢測兩峰略有重疊,基本滿足樣品檢測的要求。保健食品方法的萃取方法中用pH= 3的磷酸水溶液提取過程中要將部分酸式MK轉化成內酯式,這導致酸式含量減少,并且提取不夠徹底,過程步驟復雜,隨機誤差偏大;臺灣方法的萃取時間較短,無法將MK萃取完全,導致MK總含量偏低,并且出峰時間過早,尤其酸式MK的檢測峰與雜質峰發生重疊,影響檢測結果的準確性;朱華等的方法萃取效率最高,液相檢測峰型和出峰時間都比較理想,能夠比較準確的反應其出紅曲粉中的存在形態和含量。
4 結語
通過比較輕工方法、保健食品方法、臺灣方法以及朱華等的檢測MK的方法,發現,采用不同的方法(主要是目標物的萃取條件和色譜條件),對同一樣品會得到不同的檢測結果。輕工方法中的提取條件比較符合樣品檢測要求,但在該方法的色譜條件中,酸式和內酯式MK兩峰易出現重疊,導致檢測誤差;保健食品方法中的樣品提取過程中,需要將部分酸式MK轉化成內酯式,導致檢測結果中酸式比例偏低,且操作過程步驟復雜,且該法只用內酯式的MK表示含量,對同時含有酸式和內酯式MK的紅曲產品有失公允;臺灣方法中的樣品提取時間較短,會導致MK提取不夠徹底,并且HPLC出峰過早,易使雜質峰重疊;朱華等的檢測方法中樣品提取和色譜條件比較符合樣品檢測的要求,過程簡便快捷,具有較高的實用價值。
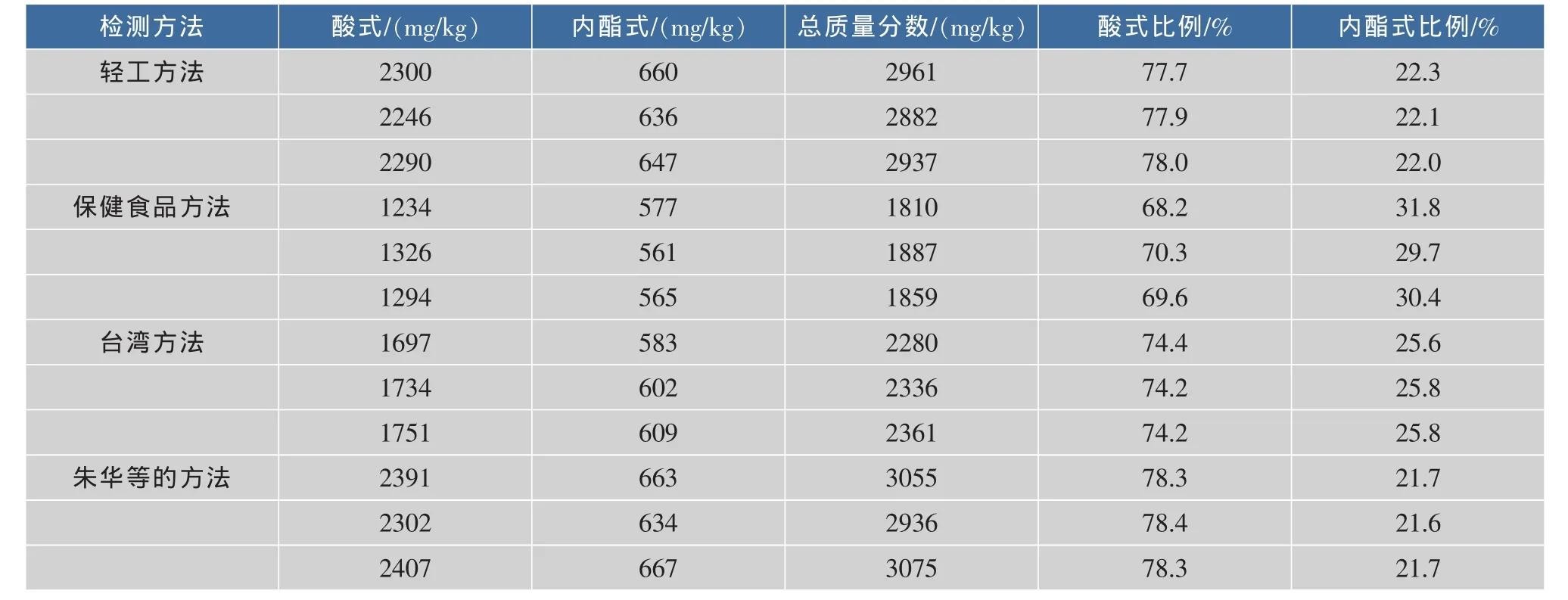
表2 不同提取方法結果比較Table 2 Compares the results of different extraction methods
作者研究的HPLC檢測方法只采用了一種柱子對不同方法所獲得的提取液進行檢測,難免對其它方法中涉及到HPLC的有關條件無法完全模擬或滿足,因此也不能完全否認這些方法。
[1]AKIRA Endo.MK,a new hypocholesterolemic agent produced by a Monascusspesies[J].J Antibiotics,1979,32:852-854.
[2]OUYANG Shu,ZHU Baoquan,GONG Bingyong.Study of cholesterol biological synthetase inhibitors-antibiotics SIPI-89-17-III [J].Journal of Chinese Antibiotics,1998,18(2):128-135.
[3]YANG Dajin,FANG Congrong,MA Lan,et al.Study on determination method of lovastatin in health foods[J].Chinese Journal of Food Hygiene,2003,15(2):125-127.
[4]CHEN Yun,CHEN Ye,XU Ganrong.The effect of dring and radiation on the Monacolin K content in functional monascus red rice[J].Journal of Food Science and Biotechnology,2008,27(4):16-19.
[5]中華人民共和國國家發展和改革委員會.中華人民共和國輕工行業標準[S].QB/T 2847-2007.
[6]中華人民共和國衛生部.保健食品檢驗與評價技術規范[S].GB 16740-2003.
[7]臺灣地區“行政院”衛生署.食品中MK之檢驗方法[S].1011900494-2012.
[8]ZHU Hua,XU Ganrong,CHEN Yun.HPLC analysis of acid form and lactone form Monacolin K[J].Journal of Wuxi University of Light Industry,2003,22(3):46-52.
[9]United States Pharmacopeial Convention United States Pharmacopeial Convention.Lovastatin and Lovastatin tablets[M].USP35,US,2012.2850-2851.
Compare of Monacolin K Detection Method in Lipid-Lowering Monascus Products
ZHAO Guanglong, ZHANG Bobo, XU Ganrong*
(School of Biotechnology,Jiangnan University,Wuxi 214122,China)
To accurately examine the amounts of Monacolin K (MK)in lipid-reducing Monascus products,in this paper we compared to the light industry standard of the People's Republic of China(2007),the health care food inspection and evaluation specification(2003),Taiwan food inspection method of MK (2012)and Zhu Hua et al.applied method.The results show that Zhu Hua et al. applied method was superior.MK linearity was good within 10~400 μg/mL,and addition recovery rate was 96.3%,RSD=1.6%.This method was simple,quick and accurate to examine the amounts of MK,with very great practical value.
lipid-lowering monascus,MK,detection method
R 927.11
A
1673—1689(2017)01—0046—05
2015-03-20
國家“十二五”科技支撐計劃項目(2011BAD23B02)。
*通信作者:許贛榮(1957—)男,江蘇無錫人,工學博士,教授,主要從事功能性紅曲及微生物發酵法生產天然色素研究。
E-mail:grxu123@126.com
趙光隆,張薄博,許贛榮.降脂紅曲產品中Monacolin K檢測方法的比較[J].食品與生物技術學報,2017,36(01):46-50.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
