AT1R-CaN信號通路在乳鼠肥大心室肌細胞Nav1.5蛋白表達調控中的作用*
2017-02-28 02:49:28夏桂玲何炯紅
中國病理生理雜志 2017年2期
鄧 娜, 夏桂玲, 楊 龍, 何炯紅, 李 雋, 田 銀, 楊 英
(貴州醫科大學附屬人民醫院心內科,貴州 貴陽 550002)
AT1R-CaN信號通路在乳鼠肥大心室肌細胞Nav1.5蛋白表達調控中的作用*
鄧 娜, 夏桂玲, 楊 龍△, 何炯紅, 李 雋, 田 銀, 楊 英
(貴州醫科大學附屬人民醫院心內科,貴州 貴陽 550002)
目的: 探討血管緊張素Ⅱ 1型受體(AT1R)-鈣調神經磷酸酶(CaN)信號通路在乳鼠肥大心室肌細胞Nav1.5 mRNA和蛋白表達調控中的作用。方法: 分離1日齡SD乳大鼠心室獲心室肌細胞,分為對照(control)組、苯腎上腺素(PE)組、 氯沙坦(Los)+PE組和環孢素A(CsA)+PE組;重組腺病毒shRNA干擾載體介導CaN A亞基β亞型(CnAβ)基因沉默分為腺病毒空載體(Ad-Null)組、Ad-Null+PE組、重組腺病毒CnAβshRNA1(Ad-CnAβshRNA1)組和Ad-CnAβshRNA1+PE組。實時熒光定量逆轉錄PCR檢測腦鈉尿肽(BNP)、β-肌球蛋白重鏈(β-MHC)和Nav1.5的mRNA表達。Western blot法檢測全細胞提取蛋白CnAβ和Nav1.5的表達。結果: PE干預24 h明顯增加心室肌細胞蛋白/DNA比值、細胞BNP和β-MHC的mRNA表達以及細胞面積;上調CnAβ蛋白表達,下調Nav1.5蛋白表達。CsA和Los干預明顯抑制PE干預的上述效應。PE下調Nav1.5的mRNA表達,但Los和CsA不能抑制此種效應。Ad-CnAβshRNA1沉默乳鼠心室肌細胞CnAβ基因抑制了PE對BNPmRNA的上調作用,抑制了PE對Nav1.5蛋白表達的下調作用。結論:AT1R-CaN信號通路參與調控培養的乳鼠肥大心室肌細胞Nav1.5蛋白表達的調控。
心肌肥大; 室性心律失常; 鈉離子通道; 血管緊張素Ⅱ 1型受體; 鈣調神經磷酸酶
心臟肥大是心臟在長期超負荷下的一種有效代償,但終因心室重構失代償而發展為充血性心力衰竭(congestive heart failure,CHF)[1]。心臟性猝死(sudden cardiac death,SCD)是CHF患者主要的死亡原因[2],超過85%的嚴重CHF患者體表心電圖記錄到室性心律失常[3]。心室動作電位由一系列離子通道所產生的離子流形成。心室動作電位時程(action potential duration,APD)延長是心衰的共同特征[4],由此產生興奮性恢復延遲,易發早期和晚期后除極,還將進一步誘發沖動傳導和折返異常[5],即心室電重構。離子通道是產生動作電位的分子基礎,離子通道重構是心室電重構的重要基礎。
鈉離子通道是由結構蛋白α亞單位Nav1.5、輔助蛋白β亞單位以及一些調節性蛋白所構成的大分子復合體,其中Nav1.5是完成通道功能的主要結構,可引起心肌細胞動作電位的快速上升,同時使沖動在心肌組織間快速傳導[6],鈉離子通道的這些功能特點,使其在室性心律失常相關的SCD中起到了重要作用[7]。
研究表明血管緊張素Ⅱ 1型受體(angiotensin Ⅱ type 1 receptor,AT1R)和鈣調神經磷酸酶(calcineurin,CaN)分別通過一定途徑參與了心肌肥大及肥大心肌離子通道重構[8-9],而且CaN作為AT1R的下游信號分子促進心肌肥大的發生發展[10],但AT1R是否通過CaN參與肥大心室肌細胞鈉離子通道重構的調控尚不明確。本研究旨在通過體外培養原代心室肌細胞,探討AT1R-CaN信號通路對肥大心室肌細胞鈉離子通道重構的調控作用。
材 料 和 方 法
1 主要試劑與儀器
高糖DMEM培養基、特優級胎牛血清(fetal bovine serum,FBS)、胰蛋白酶、II型膠原酶和5-溴脫氧尿嘧啶核苷(5-bromo-2’-deoxyuridine,BrdU)均為Gibco產品;介導沉默CaN A亞基β亞型(CnAβ)基因的重組腺病毒shRNA干擾載體 (Ad-CnAβshRNA)及空載體(Ad-Null)由漢恒生物科技有限公司包裝制備;苯腎上腺素(phenylephrine, PE)、環孢素A(cyclosporin A, CsA)和氯沙坦(losartan,Los)均為Sigma產品;兔抗大鼠α-橫紋肌肌動蛋白(alpha-sarcomeric actin,α-SCA)抗體、山羊抗兔FITC-IgG和BCA蛋白質定量試劑盒購自北京中杉金橋公司;DNA/RNA/蛋白質分離試劑盒為Omega產品;兔源Nav1.5抗體購自CST;兔源CnAβ抗體購自Merck Millipore;TransScript?First-Strand cDNA Synthesis SuperMix和TransStart? Top Green qPCR SuperMix購自北京全式金生物技術有限公司;其余試劑均為進口分裝或國產分析純。恒溫磁力攪拌器(金壇市科析儀器有限公司);Bio-Rad 550酶標儀、Bio-Rad化學發光儀(Bio-Rad);倒置顯微鏡(Olympus);熒光正置顯微鏡(Leica);PCR儀(MJ Research)。
2 實驗動物
1日齡SD乳大鼠,雌雄不限,清潔級別,由北京大學醫學部動物中心提供,許可證號為SYXK(京)2011-0039。
3 實驗方法
3.1 乳鼠心室肌細胞分離與培養 分離乳鼠左心室,保留室間隔,酶解法分離細胞,差速貼壁與BrdU結合純化獲得心室肌細胞。
3.2 心室肌細胞的鑒定 細胞爬片培養48 h,免疫熒光法檢測α-SCA抗原。以乳鼠心臟成纖維細胞作為陰性對照。
3.3 最佳Ad-CnAβshRNA的篩選 原代心室肌細胞培養48 h,以1×106個細胞數計算,選取MOI=50對應病毒量的 Ad-CnAβshRNA1、Ad-CnAβshRNA2、Ad-CnAβshRNA3及Ad-Null,感染心室肌細胞48 h,免疫印跡檢測各組心室肌細胞CnAβ蛋白表達情況,CnAβ蛋白表達量最低所對應的Ad-CnAβshRNA為最佳Ad-CnAβshRNA。結果顯示Ad-CnAβshRNA1感染心室肌細胞48 h CnAβ蛋白表達下降最為顯著,用于后續實驗。
3.4 細胞干預分組 (1) 試劑干預:細胞培養48 h,更換無血清DMEM培養基,分組干預。對照組細胞不予干預,共培養26 h;PE組細胞培養2 h后給予PE 100 μmol/L,干預24 h;Los+PE組細胞給予氯沙坦10 μmol/L預處理2 h,繼之加入PE 100 μmol/L干預24 h;CsA+PE組細胞給予環孢素10 μg/L預處理2 h,繼之加入PE 100 μmol/L干預24 h。(2) 基因沉默干預:細胞培養48 h,更換成無血清無雙抗DMEM培養基,分組干預。Ad-Null:加入MOI=50對應量的腺病毒空載體,感染6 h后更換成2倍體積新鮮無血清無雙抗DMEM,繼續培養至48 h。Ad-Null+PE組:加入MOI=50對應量的腺病毒空載體,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,感染24 h后加PE 100 μmol/L,繼續培養至48 h。Ad-CnAβshRNA1組:加入MOI=50對應量的具有最佳干擾效果的Ad-CnAβshRNA1,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,繼續培養至48 h。Ad-CnAβshRNA1+PE組:加入MOI=50對應的Ad-CnAβshRNA1,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,轉染24 h后加PE 100 μmol/L,繼續培養至48 h。
3.5 肥大刺激有效性鑒定 按照Omega DNA/RNA/蛋白質分離試劑盒說明測定共提取細胞DNA和蛋白,并分別測其含量,計算蛋白/DNA比值。RT-qPCR測定心室肌細胞腦鈉尿肽(brain natriuretic peptide, BNP)和β-肌球蛋白重鏈(β-myosin heavy chain, β-MHC)的mRNA表達。結晶紫染色拍照,軟件測量細胞面積大小。
3.6 RT-qPCR檢測mRNA水平 利用Oligo 6.0軟件設計引物(序列見表1)。TRIzol法提取細胞總mRNA,按TransScript?First-Strand cDNA Synthesis SuperMix和TransStart?Top Green qPCR SuperMix說明書的步驟檢測并計算各基因的mRNA相對表達量。

表1 各基因的引物序列
3.7 Western blot實驗 提取細胞總蛋白,測定蛋白濃度。取40 μg總蛋白上樣,電泳后轉移蛋白至NC膜上,5%脫脂牛奶封閉1 h。加兔抗大鼠GAPDH抗體(1∶1 000),兔抗大鼠Nav1.5抗體(1∶1 000)或兔抗大鼠CnAβ抗體(1∶500),4 ℃孵育過夜。充分漂洗后,加入辣根過氧化酶標記的 II 抗室溫孵育1 h。充分漂洗后顯影、攝片并進行條帶灰度定量測定。
4 統計學處理
采用Graphpad Prism 6軟件進行統計分析。數據采用均數±標準差(mean±SD)表示。應用單因素方差分析(one-way ANOVA)及Tukey檢驗進行多組間兩兩比較,以P<0.05為差異有統計學意義。
結 果
1 原代心室肌細胞的鑒定
細胞培養48 h,倒置顯微鏡下可見心室肌細胞呈長梭形、多邊形或不規則形,核仁清晰可見,單個細胞貼壁后可見節律性舒縮搏動。α-SCA抗體免疫熒光染色心室肌細胞陽性,成纖維細胞陰性,見圖1。
2 肥大刺激有效性鑒定
給予PE刺激24 h,心室肌細胞蛋白/DNA比值增大,BNP和β-MHC的mRNA表達明顯上調,心室肌細胞表面積增大。該結果提示PE刺激心室肌細胞肥大。CsA和Los干預抑制PE引起的上述效應,見圖2~4。
3 Los和CsA抑制了PE刺激增加的心室肌細胞CnAβ的蛋白表達
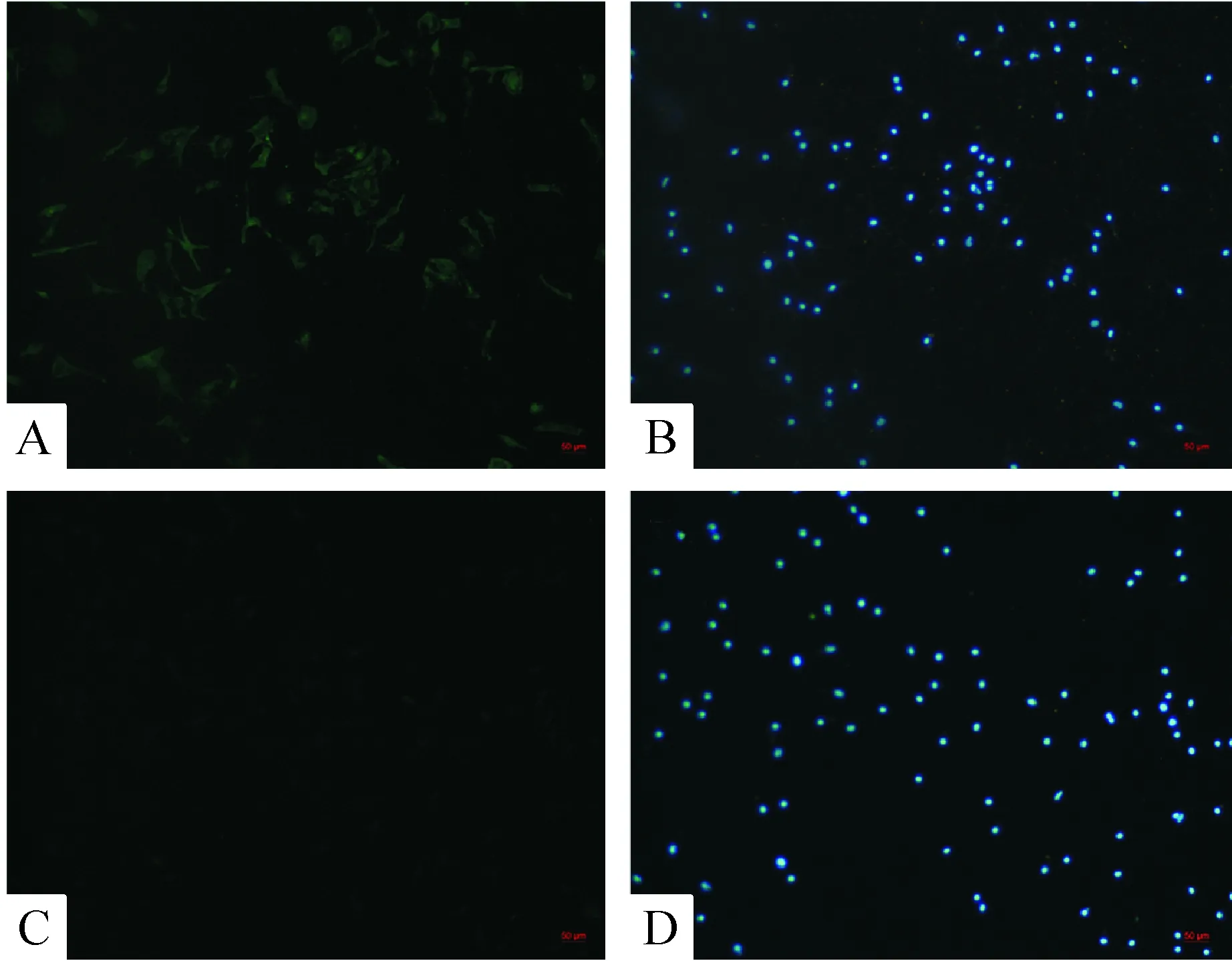
Figure 1.α-Sarcometric actin (α-SCA) immunofluorescence staining of ventricular myocytes and fibroblast (×100). A: ventricular myocytes with α-SCA immunofluorescence staining; B: ventricular myocyte nuclei with DAPI staining; C: cardiac fibroblasts with α-SCA immunofluorescence staining; D: cardiac fibroblast nuclei with DAPI staining.
圖1 心室肌細胞和成纖維細胞α-橫紋肌肌動蛋白免疫熒光染色
給予PE刺激心室肌細胞24 h,細胞CnAβ蛋白表達明顯上調;Los和CsA顯著抑制了PE的此種效應,見圖5。
4 Los和CsA對PE刺激Nav1.5的mRNA和蛋白表達的調控作用
給予PE刺激心室肌細胞24 h,細胞Nav1.5的mRNA和蛋白表達明顯下調; Los和CsA抑制PE刺激的Nav1.5蛋白表達下調,但未能抑制PE誘導的Nav1.5 mRNA表達下調,見圖6。
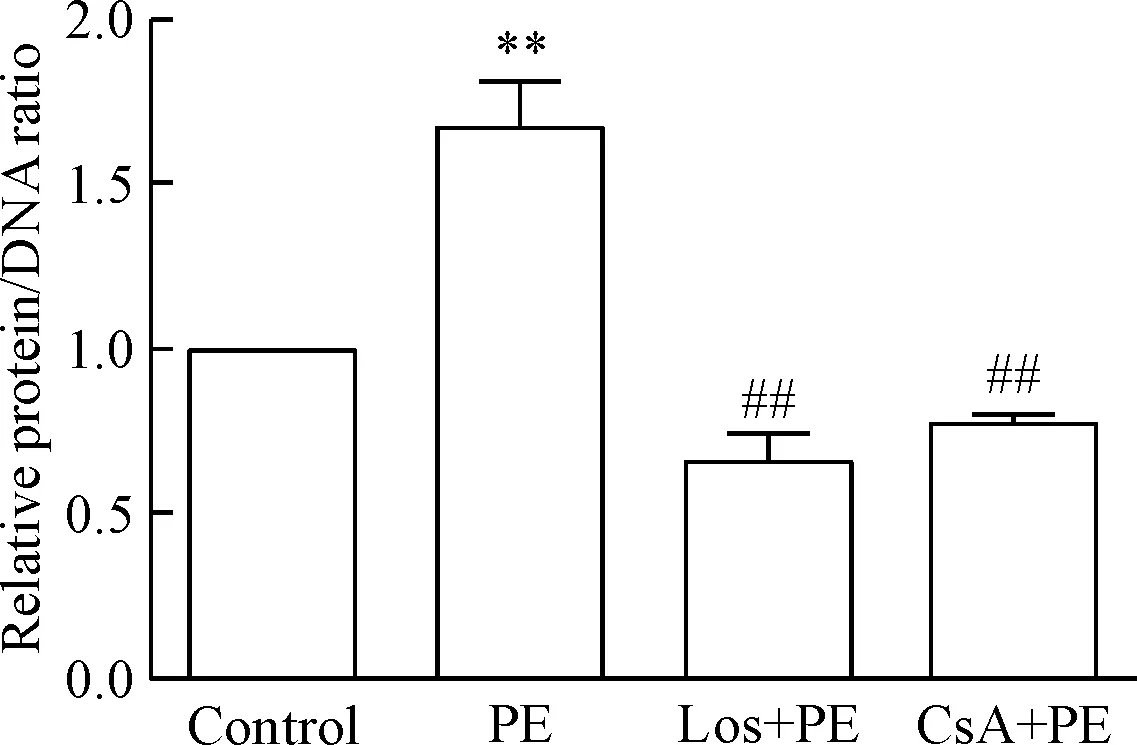
Figure 2.Pretreatment of neonatal rat ventricular myocytes with Los or CsA blocked the ability of PE to increase the protein-to-DNA ratio. Mean±SD.n=4.**P<0.01vscontrol group;##P<0.01vsPE group.
圖2 Los和CsA抑制PE刺激增加的心室肌細胞蛋白/DNA比值
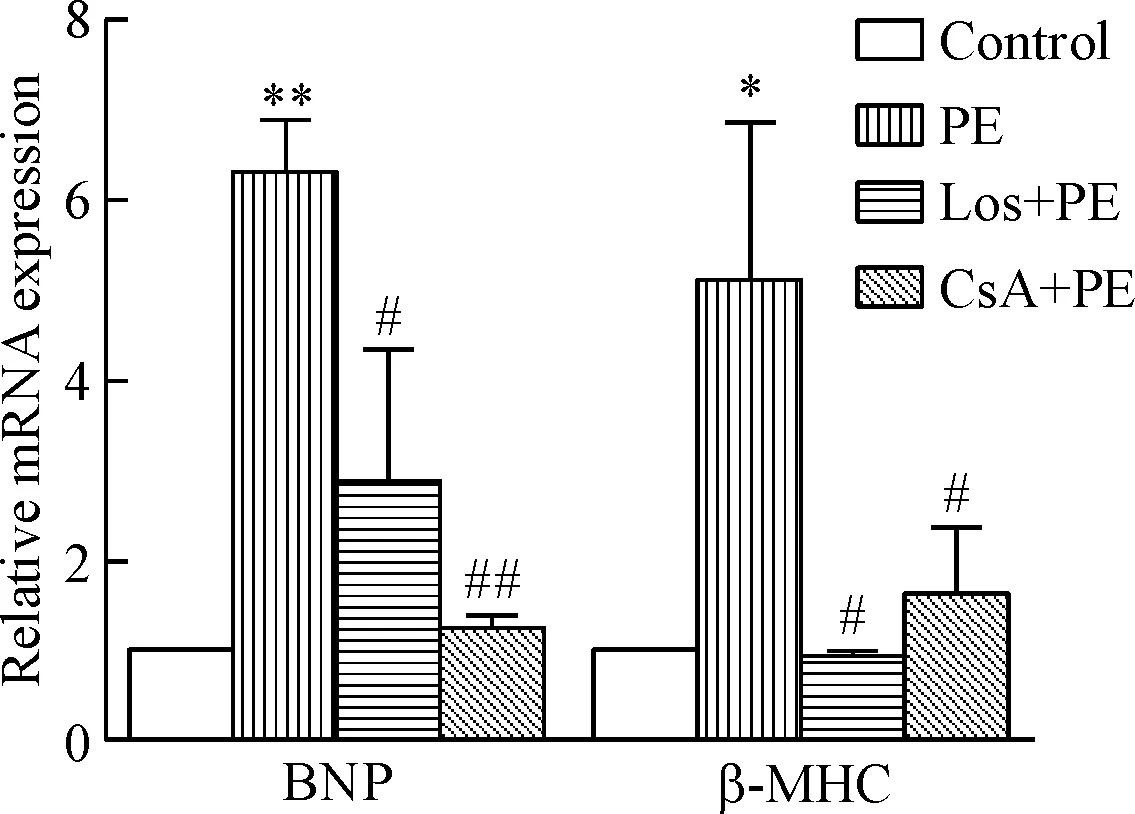
Figure 3.Pretreatment of neonatal rat ventricular myocytes with Los or CsA blocked the ability of PE to increase the mRNA expression of BNP and β-MHC. Mean±SD.n=3.*P<0.05,**P<0.01vscontrol group;#P<0.05,##P<0.01vsPE group.
圖3 Los和CsA抑制PE刺激增加的心室肌細胞肥大基因表達
5 Ad-CnAβshRNA1干擾CnAβ表達抑制PE增加的BNP mRNA表達
心室肌細胞培養48 h,Ad-CnAβshRNA1和Ad-Null分別感染心室肌細胞24 h后給予PE刺激24 h。結果顯示PE刺激感染Ad-Null的心室肌細胞,其BNP的mRNA表達明顯上調;而在感染Ad-CnAβshRNA1的心室肌細胞,PE刺激不能使細胞BNP的mRNA表達上調,見圖7。
6 Ad-CnAβshRNA1干擾CnAβ表達對PE刺激Nav1.5的蛋白表達的調控作用
心室肌細胞培養48 h,Ad-CnAβshRNA1和Ad-Null分別感染心室肌細胞24 h后給予PE刺激24 h。結果顯示感染Ad-Null的細胞Nav1.5蛋白表達明顯下調;感染Ad-CnAβshRNA1的細胞Nav1.5蛋白表達無明顯下調,見圖8。
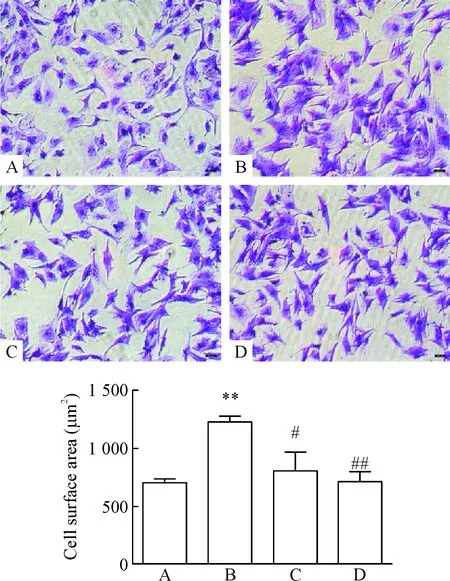
Figure 4.Pretreatment of neonatal rat ventricular myocytes with Los or CsA blocked the ability of PE to increase the size of the cell surface (crystal violet staining, ×100). A: control group; B: PE group; C: Los+PE group; D: CsA+PE group. Mean±SD.n=3.**P<0.01vscontrol group;#P<0.05,##P<0.01vsPE group.
圖4 Los和CsA抑制PE刺激增加的心室肌細胞表面積
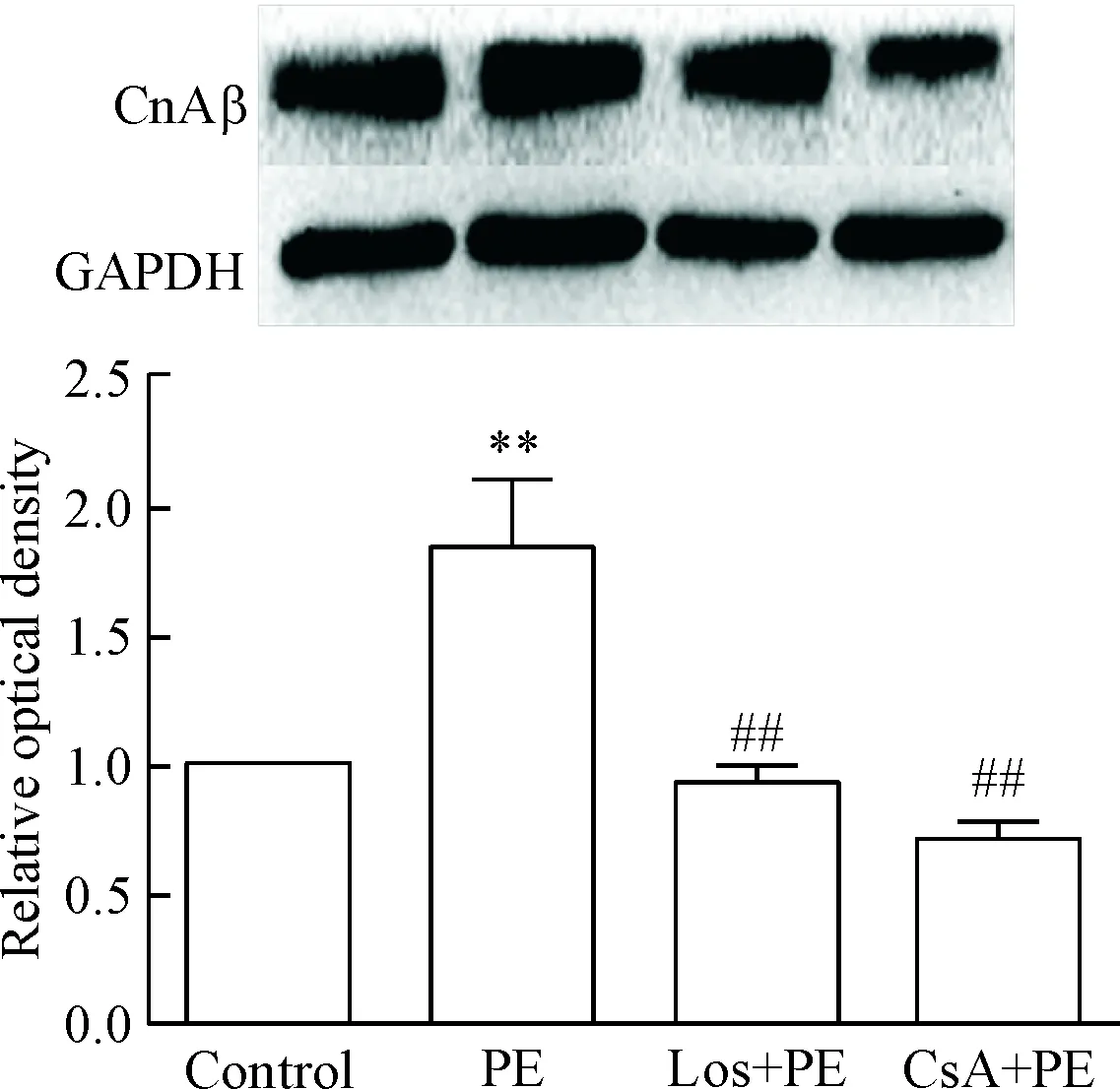
Figure 5.Pretreatment of neonatal rat ventricular myocytes with Los or CsA blocked the ability of PE to increase protein expression of CnAβ. Mean±SD.n=5.**P<0.01vscontrol group;##P<0.01vsPE group.
圖5 Los和CsA抑制PE刺激增加的心室肌細胞CnAβ的蛋白表達
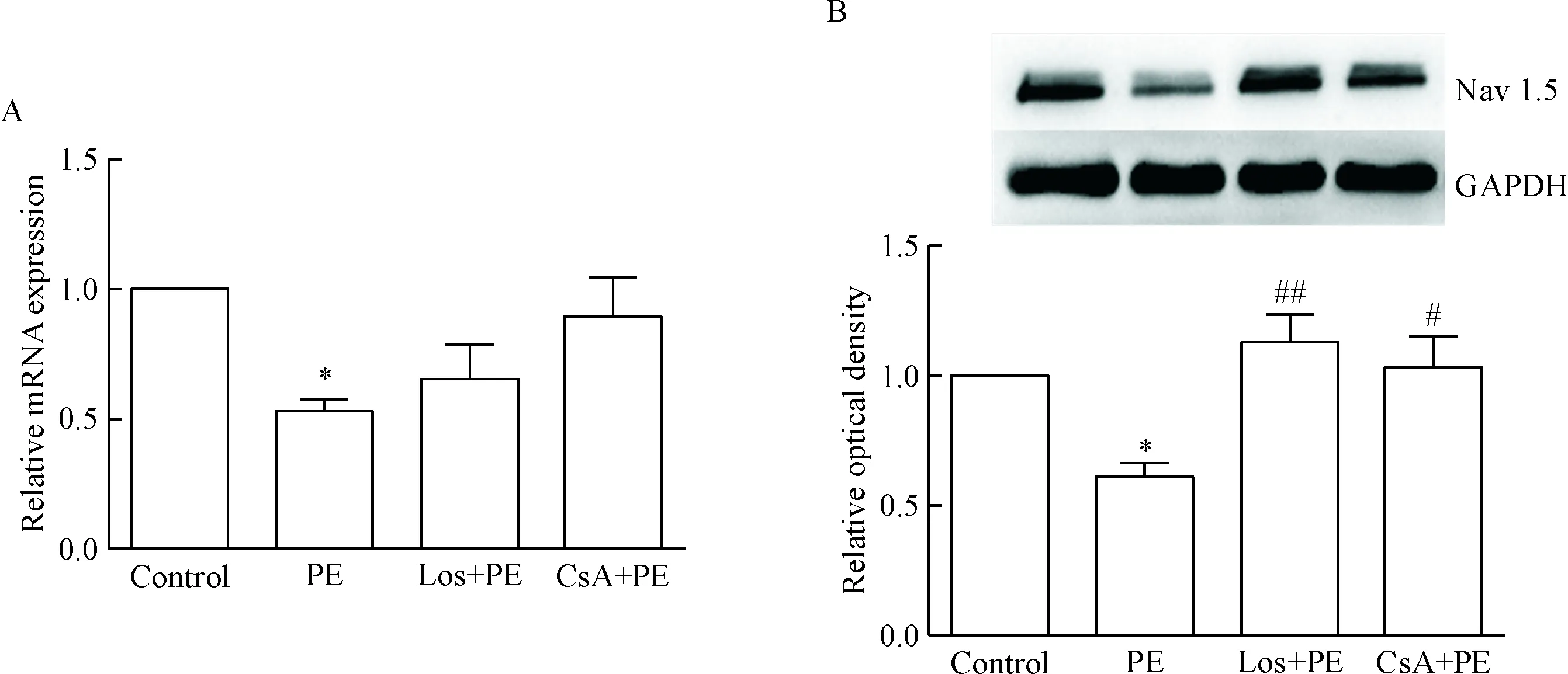
Figure 6.Los and CsA both increased the mRNA (A;n=3) and protein (B;n=5) expression of Nav1.5 reduced by PE treatment. Mean±SD.*P<0.05vscontrol group;#P<0.05,##P<0.01vsPE group.
圖6 Los和CsA增加PE所抑制的Nav1.5蛋白表達
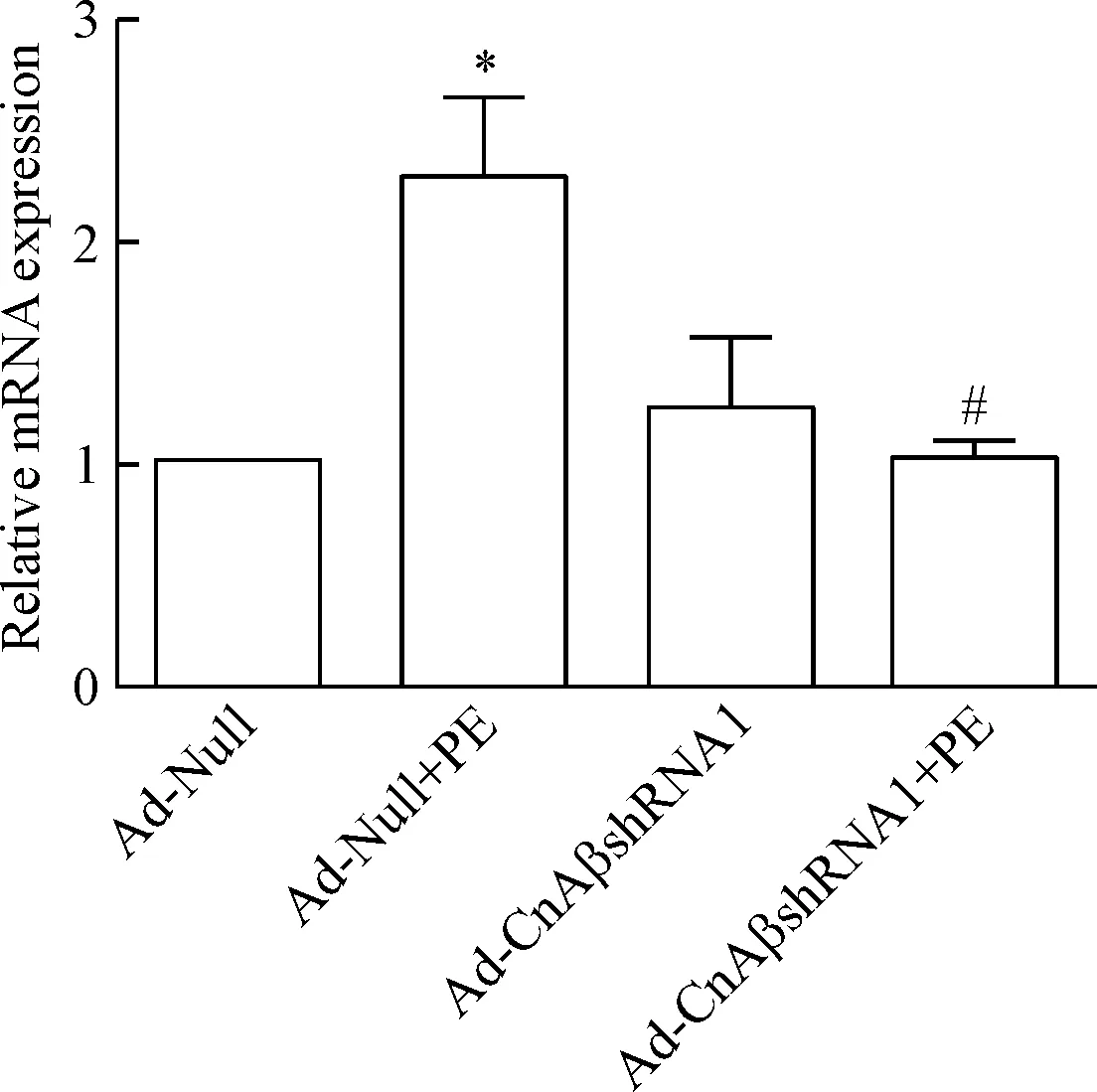
Figure 7.Knockdown ofCnAβexpression in the neonatal rat ventricular myocytes using Ad-CnAβshRNA1 inhibited the ability of PE to increase the mRNA expression of BNP. The data was presented by relative value of Ad-Null group. Mean±SD.n=3.*P<0.05vsAd-Null group;#P<0.05vsAd-Null+PE group.
圖7 Ad-CnAβshRNA1敲減CnAβ表達抑制PE增加BNP mRNA表達的效應
討 論
心肌肥大、心衰患者室性心律失常風險顯著增加,其機制之一是存在心室電重構,如心室動作電位時程、易發早期和晚期后除極、沖動傳導和折返異常等[5]。離子通道重構是這些心臟電生理變化的重要基礎。
α1-腎上腺素受體(α1-adrenergic receptors,α1-ARs)屬于G蛋白偶聯受體(G protein-coupled receptor,GPCRs),是腎上腺素、去甲腎上腺素等兒茶酚胺類心臟肥大效應的重要調節因素[11]。本研究結果顯示,給予PE干預24h的原代心室肌細胞其蛋白/DNA比值明顯增高,BNP和β-MHC的mRNA表達上調,心室肌細胞表面積顯著增加,均提示PE誘導了心室肌細胞肥大[12]。肥大因素刺激心肌局部腎素-血管緊張素系統(renin-angiotensin system,RAS)被激活,細胞內AT1R活化,參與了促心肌肥大過程。CaN是AT1R下游的信號分子之一,在心肌肥大的發生發展過程中起著重要作用[13]。本研究使用PE誘導心室肌細胞肥大,再分別用AT1R阻斷劑Los、CaN抑制劑CsA和Ad-CnAβshRNA特異性沉默心室肌細胞CnAβ基因表達,阻斷AT1R-CaN信號通路不同位點,均明顯抑制了PE的促肥大效應,說明AT1R-CaN信號通路參與PE誘導的乳大鼠心室肌細胞肥大。但是AT1R-CaN信號通路是否參與肥大心室肌細胞中離子通道重構,未見相關報道。
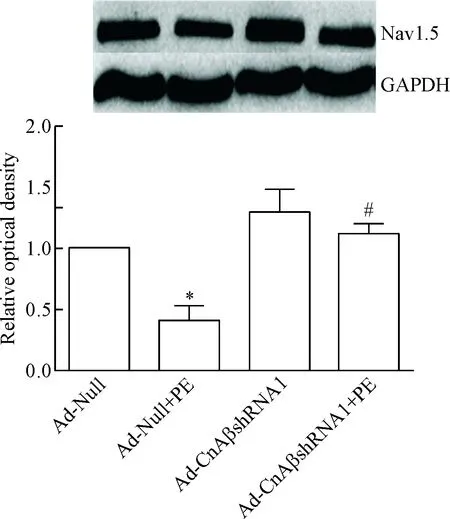
Figure 8.Knockdown ofCnAβexpression in the neonatal rat ventricular myocytes using Ad-CnAβshRNA1 inhibited the ability of PE to reduce protein expression of Nav1.5. Mean±SD.n=5.*P<0.05vsAd-Null group;#P<0.05vsAd-Null+PE group.
圖8 Ad-CnAβshRNA1敲減CnAβ表達增加PE所抑制的Nav1.5蛋白表達
鈉離子通道是心肌細胞快速去極化的主要組成成分,形成動作電位上升支,隨后引發興奮收縮偶聯級聯[14]。研究發現心衰患者除動作電位時程改變外,心肌傳導異常也是SCD風險增高的主要原因[15]。鈉離子通道的活性是心室肌細胞興奮傳導的決定因素之一。鈉離子通道的丟失能引起興奮傳導失敗或傳導減慢,導致心衰心肌離散度增強從而促進折返和室性心律失常。本研究PE可致肥大心室肌細胞鈉離子通道亞基Nav1.5的mRNA和蛋白表達下調,意味著PE誘導的心室肌肥大存在鈉離子通道丟失,可能導致肥大心室肌細胞中鈉離子通道功能減弱,繼而快速鈉電流減小,引起心室肌細胞興奮傳導異常等電重構現象及興奮收縮偶聯的異常。這在一定程度上解釋了心衰患者易發SCD與Nav1.5表達異常引起心室電重構有關。本研究Los、CsA和Ad-CnAβshRNA特異性沉默心室肌細胞CnAβ基因表達均在蛋白水平抑制PE的上述效應,然而我們的研究并未發現分別阻斷AT1R和CaN能抑制PE對肥大心室肌細胞Nav1.5 mRNA表達的影響,說明AT1R-CaN信號通路在蛋白水平調控PE對心室肌細胞Nav1.5的影響,可能是通過影響Nav1.5的翻譯效率或Nav1.5的穩定性,從而影響Nav1.5的合成或降解,這將在今后的實驗中進一步驗證。近期的研究表明在過表達AT1R的轉基因小鼠中心室肌鈉通道表達下調,鈉離子流密度下降了60%,動作電位上升的最大速度減慢,并導致了QRS波時限增寬,均支持我們的上述研究結果[15]。
綜上所述,AT1R-CaN信號通路參與PE誘導的乳鼠肥大心室肌細胞Nav1.5蛋白表達的調控。
[1] Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy[J]. J Mol Cell Cardiol, 2016, 97(2): 245-262.
[2] Mareev VY, Mareev YV. Methods of prevention of sudden death in chronic heart failure[J]. Kardiologiia, 2015, 55(9): 72-83.
[3] Lane RE, Cowie MR, Chow AW. Prediction and prevention of sudden cardiac death in heart failure[J]. Heart, 2005, 91(5):674-680.
[4] Wang Y, Hill JA. Electrophysiological remodeling in heart failure[J]. J Mol Cell Cardiol, 2010, 48(4):619-632.
[5] Wang Y, Tandan S, Hill JA. Calcineurin-dependent ion channel regulation in heart[J]. Trends Cardiovasc Med, 2014, 24(1):14-22.
[6] Bezzina CR, Rook MB, Groenewegen WA, et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system[J]. Circ Res, 2003, 92(2):159-168.
[7] Antzelevitch C. Brugada syndrome[J]. Pacing Clin Electrophysiol, 2006, 29(10): 1130-1159.
[8] Zhou C, Ziegler C, Birder LA, et al. Angiotensin II and stretch activate NADPH oxidase to destabilize cardiac Kv4.3 channel mRNA[J]. Circ Res, 2006, 98(8):1040-1047.
[9] Huo R, Sheng Y, Guo WT, et al. The potential role of Kv4.3 K+channel in heart hypertrophy[J]. Channels, 2014, 8(3):203-209.
[10]Rohini A, Agrawal N, Koyani CN, et al. Molecular targets and regulators of cardiac hypertrophy[J]. Pharmacol Res, 2010, 61(4):269-280.
[11]Cotecchia S, Del Vescovo CD, Colella M, et al. The α1-adrenergic receptors in cardiac hypertrophy: signaling mechanisms and functional implications[J]. Cell Signalling, 2015, 27(10):1984-1993.
[12]Huang S, Zou X, Zhu JN, et al. Attenuation of micro-RNA-16 derepresses the cyclins D1, D2 and E1 to provoke cardiomyocyte hypertrophy[J]. J Cell Mol Med, 2015, 19(3): 608-619.
[13]Wu X, Zhang T, Bossuyt J, et al. Local InsP3-dependent perinuclear Ca2+signaling in cardiac myocyte excitation-transcription coupling[J]. J Clin Invest, 2006, 116(3): 675-682.
[14]Zaklyazminskaya E, Dzemeshkevich S. The role of mutations in theSCN5Agene in cardiomyopathies[J]. Biochim Biophys Acta, 2016, 1863(7 Pt B):1799-1805.
[15]Mathieu S, El Khoury N, Rivard K, et al. Reduction in Na+current by angiotensin II is mediated by PKCalpha in mouse and human-induced pluripotent stem cell-derived cardiomyocytes[J]. Heart Rhythm, 2016, 13(6):1346-1354.
(責任編輯: 林白霜, 羅 森)
RoleofAT1R-CaNsignalingpathwayinregulationofNav1.5proteinexpressioninhypertrophicventricularmyocytesfromneonatalrats
DENGNa,XIAGui-ling,YANGLong,HEJiong-hong,LIJun,TIANYin,YANGYing
(DepartmentofCardiology,GuizhouProvincialPeople’sHospital,ThePeople’sHospitalofGuizhouMedicalUniversity,Guiyang550002,China.E-mail:yanglong1001@163.com)
AIM: To investigate the effect of angiotensin II type 1 receptor (AT1R)-calcineurin (CaN) signaling pathway on the expression of sodium current channel Nav1.5 at mRNA and protein levels in the hypertrophic ventricular myocytes from neonatal rats. METHODS: The ventricular myocytes were isolated from the ventricles of 1-day-old neonatal Sprague-Dawley rats and were divided into 4 groups according to different drug intervention as control group, phenylephrine (PE) group, losartan (Los)+PE group and cyclosporin A (CsA)+PE group. The method of RNA interference mediated by adenovirus carrying short hairpin RNA (shRNA) was used to knock down the gene which encodes the beta subtype of CaN A subunit (CnAβ) and the cells were divided into 4 groups as Ad-Null group, Ad-Null+PE group, Ad-CnAβshRNA1 group and Ad-CnAβshRNA1+PE group. The mRNA expression of brain natriuretic peptide (BNP), β-myosin heavy chain (β-MHC) and Nav1.5 was detected by RT-qPCR. The protein levels of CnAβ and Nav1.5 in the whole-cell extracts were determined by Western blot analysis. RESULTS: Treatment of the neonatal rat ventricular myocytes with PE for 24 h increased the protein-to-DNA ratio and the mRNA expression of BNP and β-MHC. The size of the cell surface was also increased after PE treatment. Treatment of the cells with PE increased the protein expression of CnAβ, and reduced the protein expression of Nav1.5. Both Los and CsA prevented those effects of PE. The mRNA expression of Nav1.5 was reduced by PE, and no significant difference of Nav1.5 mRNA expression among PE group, Los+PE group and CsA+PE group was observed. Silencing ofCnAβin the neonatal rat ventricular myocytes using Ad-CnAβshRNA1 inhibited the ability of PE to increase the mRNA expression of BNP, and diminished the ability of PE to reduce the protein expression of Nav1.5. CONCLUSION: AT1R-CaN signaling pathway participates in regulating protein expression of Nav1.5 in the hypertrophic ventricular myocytes from neonatal rats.
Cardiac hypertrophy; Ventricular arrhymaias; Sodium channels; Angiotensin II type 1 receptor; Calcineurin
1000- 4718(2017)02- 0221- 06
2016- 08- 02
2016- 12- 01
國家自然科學基金資助項目(No. 81260040);貴州省科學技術基金資助項目(黔科合J字[2012]2239號)
R
A
10.3969/j.issn.1000- 4718.2017.02.005
雜志網址: http://www.cjpp.net
△通訊作者 Tel: 0851-85937194; E-mail: yanglong1001@163.com
