間歇低氧后正常氧培養(yǎng)不同時間胎鼠海馬神經(jīng)元HMGB1、NMDA表達變化
2017-02-23 08:32:00孫文文吳丹韓笑宋毅軍馮靖曹潔
山東醫(yī)藥 2017年2期
關(guān)鍵詞:海馬
孫文文,吳丹,韓笑,宋毅軍,馮靖,曹潔
(1天津醫(yī)科大學研究生院,天津 300070;2天津醫(yī)科大學總醫(yī)院神經(jīng)病研究所;3天津醫(yī)科大學總醫(yī)院)
·基礎研究·
間歇低氧后正常氧培養(yǎng)不同時間胎鼠海馬神經(jīng)元HMGB1、NMDA表達變化
孫文文1,吳丹2,韓笑2,宋毅軍2,馮靖3,曹潔3
(1天津醫(yī)科大學研究生院,天津 300070;2天津醫(yī)科大學總醫(yī)院神經(jīng)病研究所;3天津醫(yī)科大學總醫(yī)院)
目的 觀察間歇低氧后正常氧培養(yǎng)不同時間胎鼠海馬神經(jīng)元高遷移率族蛋白1(HMGB1)、N-甲基-D-天冬氨酸(NMDA)表達變化。方法 孕16~18 d的SD大鼠,取其胎鼠海馬神經(jīng)元細胞培養(yǎng)至第7天,將細胞隨機分為對照組、低氧組。低氧組細胞以低氧5 min/常氧5 min進行暴露,頻率為6次/h,持續(xù)暴露時間為4 h。對照組細胞持續(xù)暴露于正常氧4 h。暴露結(jié)束后兩組細胞放入標準細胞孵育箱中繼續(xù)培養(yǎng)2、4、6、8 h。采用實時熒光定量PCR檢測兩組胎鼠海馬神經(jīng)元HMGB1、NMDA mRNA。結(jié)果 低氧組常氧培養(yǎng)2、4、6、8 h時海馬神經(jīng)元HMGB1 mRNA相對表達量分別為0.004 7±0.001 13、0.013 5±0.00 10、0.001 1±0.000 15、0.000 6±0.000 12,與對照組(0.001 7±0.000 16)相比P均<0.05,4 h時與其他各時點相比P均<0.05。低氧組常氧培養(yǎng)2、4、6、8 h時海馬神經(jīng)元NMDA mRNA相對表達量分別為0.025 7±0.002 2、0.032 2±0.001 5、0.044 3±0.011 7、0.058 3±0.009 9,與對照組(0.017 4±0.002 7)相比P均<0.05,8 h時與其他各時點相比P均<0.05。結(jié)論 胎鼠海馬神經(jīng)元間歇低氧后正常氧培養(yǎng)2、4 h時HMGB1 mRNA表達升高,6、8 h時表達降低;NMDA mRNA表達隨正常氧培養(yǎng)時間(2~8 h)延長而增加。
阻塞性睡眠呼吸暫停綜合征;間歇低氧;海馬神經(jīng)元;高遷移率族蛋白1;N-甲基-D-天冬氨酸;胎鼠
間歇低氧是阻塞性睡眠呼吸暫停綜合征(OSA)的重要病理生理特征之一[1],引起的神經(jīng)病理學改變包括神經(jīng)元死亡、認知功能障礙、情感障礙,認知功能障礙主要是由海馬CA1區(qū)域神經(jīng)元的凋亡引起[2,3]。從死亡或瀕臨死亡的細胞中釋放出來的分子可引起無菌炎癥,這些分子也被稱為損傷相關(guān)分子模式(DAMP),常見的DAMP包括熱休克蛋白、高遷移率族蛋白1(HMGB1)。N-甲基-D-天冬氨酸(NMDA)受體是一種廣泛分布于神經(jīng)突觸后膜的谷氨酸受體,其異常激活引起神經(jīng)興奮毒性,在多種神經(jīng)退行性疾病如缺血缺氧性腦損傷、阿爾茨海默病的認知損害中均有報道[4]。已有小鼠體外模型證明,慢性間歇低氧認知功能障礙與NMDA受體的過度激活、海馬神經(jīng)元細胞內(nèi)鈣超載有關(guān)[5],癲癇、神經(jīng)創(chuàng)傷等腦損傷中HMGB1可促進NMDA引起的神經(jīng)元死亡[6]。間歇低氧停止后,其誘發(fā)的神經(jīng)炎癥是否還會繼續(xù)造成神經(jīng)元損傷尚不明確。2015年12月~2016年9月,本研究觀察了胎鼠海馬神經(jīng)元間歇低氧后正常氧培養(yǎng)不同時間HMGB1、NMDA表達變化,現(xiàn)將結(jié)果報告如下。
1 材料與方法
1.1 實驗動物、試劑和儀器 健康SD大鼠購自中國人民解放軍軍事醫(yī)學科學院實驗動物中心。0.25%胰酶、胎牛血清、DMEM-F12培養(yǎng)基、Neurobasal培養(yǎng)基、B27均購自美國Gibco公司;多聚賴氨酸、谷氨酰胺購自美國Sigma公司;TRIzol試劑購自美國Invitrogen公司;HMGB1、NMDA引物合成及測序于上海生工生物公司完成;PCR試劑購自北京普凱瑞公司;混合氣購自天津泰亞氣體公司;日本Olympus光學倒置顯微鏡及圖像采集系統(tǒng);美國Bio-Rad熒光定量PCR儀。
1.2 胎鼠海馬神經(jīng)元獲取及培養(yǎng) 腹腔麻醉孕16~18 d的SD大鼠,無菌條件下取出胎鼠,并快速分離海馬組織,用眼科剪將組織剪成1 mm3的碎片,將碎片用0.125%的胰酶在37 ℃消化15 min。隨后用1∶1的種植培養(yǎng)基(1%青霉素-鏈霉素、20%胎牛血清、1%谷氨酰胺、78%DMEM/F12)終止消化。用吸管吹打制成單細胞懸液,將細胞接種到提前用多聚賴氨酸包被的培養(yǎng)皿中。4 h后將培養(yǎng)基更換為無血清培養(yǎng)基(2%B27、1%谷氨酰胺、1%青霉素-鏈霉素,96% neurobasal培養(yǎng)基),每2~3 d半量換液。海馬神經(jīng)元在37 ℃、5% CO2孵育箱中培養(yǎng)7 d。剛接種的神經(jīng)元在鏡下呈圓形,海馬神經(jīng)元培養(yǎng)4 h后細胞突起數(shù)增多,折光性很強。24 h后,大部分細胞長出3~4個突起,長度較長,有少數(shù)細胞聚合。培養(yǎng)2 d后,神經(jīng)元突起增多并延長,胞體逐漸變大。培養(yǎng)5 d后,神經(jīng)元胞體逐漸生長,突起逐漸延長,形成較密的網(wǎng)絡。培養(yǎng)7 d后,神經(jīng)元細胞生長良好,胞體清晰較大,呈三角線、圓形、棱形或多邊形,突起飽滿并形成稠密的神經(jīng)網(wǎng)絡。
1.3 胎鼠海馬神經(jīng)元分組及間歇低氧處理 采用天津醫(yī)科大學總醫(yī)院呼吸科仿真系統(tǒng)1.0設置低氧/再氧合循環(huán)模式[7]。低氧混合氣含1.5% O2、93.5% N2、5% CO2,正常氧混合氣含21% O2、74% N2、5% CO2。自行設計制作細胞培養(yǎng)艙,采用溫控器、加熱濕化器和微孔濾膜使培養(yǎng)艙中為無菌環(huán)境,溫度為37 ℃,濕度在45%~70%。海馬神經(jīng)元培養(yǎng)7 d后隨機分為低氧組和對照組。參照文獻[8],將低氧組和對照組細胞培養(yǎng)皿放在上述自制培養(yǎng)艙內(nèi),分別暴露于間歇低氧和正常氧的環(huán)境中。低氧組細胞以低氧5 min/常氧5 min進行暴露,頻率為6次/h,持續(xù)暴露時間為4 h。對照組細胞持續(xù)暴露時間是4 h。暴露結(jié)束后兩組細胞放入標準細胞孵育箱(37 ℃,5% CO2)中繼續(xù)培養(yǎng)2、4、6、8 h。
1.4 胎鼠海馬神經(jīng)元HMGB1、NMDA mRNA檢測方法 培養(yǎng)2、4、6、8 h后,棄上清,用TRIzol法提取海馬神經(jīng)元細胞總RNA(1 mL/孔)。以適量DEPC水溶解RNA,并取一部分用紫外分光光度儀測算OD260與OD280比值,并根據(jù)OD260值推算其濃度。待確定純度符合要求后,將RNA采用一步法進行real-time PCR法測定HMGB1、NMDA mRNA。HMGB1、NMDA和β-actin的基因序列均查自Gene Bank,引物序列由專業(yè)軟件Primer5.0設計。HMGB1上游引物序列:5′-ATGGGCAAAGGAGATCCTA-3′,下游引物序列:5′-ATTCATCATCATCATCTTCT-3′;NMDA上游引物序列:5′-TATGTGTGGCCTCGGATGTGT-3′,下游引物序列:5′-TCTTCCCGTGCTTGCCATT-3′;β-actin上游引物序列:5′-GAAATCGTGCGTGACATTA-3′,下游引物序列:5′-TAGGAGCCAGGGCAGTAA-3′。總反應體系20 μL,在熒光定量PCR儀(Bio-rad,美國)設定PCR循環(huán)參數(shù):42 ℃ 5 min,95 ℃ 5 min,95 ℃ 3 s,60 ℃ 25 s,72 ℃ 10 s,共40個循環(huán)。實驗結(jié)果數(shù)值以Ct值表示,每組細胞重復檢測3次,取平均值作為該組細胞的Ct值。以β-actin為內(nèi)參,應用2-ΔCt法計算HMGB1、NMDA mRNA相對表達量。
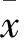
2 結(jié)果
兩組胎鼠海馬神經(jīng)元HMGB1、NMDA mRNA相對表達量比較見表1。

表1 兩組胎鼠海馬神經(jīng)元HMGB1、NMDA mRNA相對表達量比較±s)
注:與對照組相比,aP<0.05;與同組2 h時相比,bP<0.05;與同組4 h時相比,cP<0.05;與同組6 h時相比,dP<0.05。
3 討論
OSA患者的神經(jīng)認知功能隨著夜間低氧的損害程度而加重。海馬是學習記憶的主要功能腦區(qū),其中CA1區(qū)最易受缺氧損傷,間歇低氧會導致神經(jīng)元凋亡[3,9]。HMGB1是一種高度保守、分布廣泛的非組織胺DNA結(jié)合蛋白,參與多種細胞的生理功能,在細胞核調(diào)節(jié)眾多轉(zhuǎn)錄因子和DNA的相互作用。
近年來,HMGB1被定義為一種重要的促炎細胞因子,其促炎作用是在細胞外發(fā)揮的。機體受到不同因素刺激時,活化的免疫細胞主動分泌或從壞死、損傷細胞中釋放HMGB1誘發(fā)炎癥反應、缺血、免疫疾病和神經(jīng)退行性病變。HMGB1廣泛表達于不同組織,包括腦、腎等。在腦組織中,HMGB1主要分布在神經(jīng)元、星形膠質(zhì)細胞、小膠質(zhì)細胞和垂體后葉細胞,而神經(jīng)元是缺血性腦損傷釋放HMGB1的主要來源[10],可見HMGB1參與神經(jīng)炎癥反應[11]。一些酶如NADPH氧化酶、誘導型一氧化氮合酶(iNOS)和環(huán)氧化酶2(COX-2)分別通過產(chǎn)生活性氧簇、活性氮簇和前列腺素引起炎癥反應。在中樞神經(jīng)系統(tǒng)中,iNOS和COX-2分別通過產(chǎn)生活性氮簇和前列腺素引起神經(jīng)元的損傷。而在腦缺血模型中給予外源性的HMGB1可增加炎癥因子TNF-α和iNOS合成。Kim等[12]研究發(fā)現(xiàn),腦缺血后HMGB1作為促炎因子連接興奮中毒引起的急性損傷過程和延遲的炎癥過程。也有報道神經(jīng)元釋放的HMGB1在如癲癇、酗酒等情況下介導神經(jīng)炎癥[13]。細胞外HMGB1作為炎癥因子通過激活小膠質(zhì)細胞刺激其他炎癥因子的分泌,并促進與其表面受體結(jié)合,如Toll-like受體(TLR)、NF-κB受體,從而加重腦損傷,在損傷或凋亡的神經(jīng)元附近可發(fā)現(xiàn)HMGB1的累積[14,15]。在創(chuàng)傷性腦損傷中,HMGB1由損傷的神經(jīng)元細胞釋放到細胞外并激活小膠質(zhì)細胞表面的TLR4受體誘發(fā)神經(jīng)炎癥從而引起神經(jīng)元的損傷[16]。本研究發(fā)現(xiàn),胎鼠海馬神經(jīng)元間歇低氧后正常氧培養(yǎng)2、4 h海馬神經(jīng)元分泌的HMGB1 mRNA水平升高,6、8 h時表達水平降低。4 h后神經(jīng)炎癥因子分泌達到高峰并加重神經(jīng)元炎性損傷,引起神經(jīng)元死亡或凋亡數(shù)量增加,細胞分泌的HMGB1逐漸減少。表明即使間歇低氧暴露撤除,神經(jīng)元的損傷也持續(xù)存在,間歇低氧不是維持神經(jīng)炎癥的必備條件。急性損傷,例如缺氧,可以直接引起中樞神經(jīng)系統(tǒng)神經(jīng)元損傷。受損傷的神經(jīng)元通過產(chǎn)生許多有毒的內(nèi)源性物質(zhì),例如HMGB1釋放到細胞外和膠質(zhì)細胞表面的模式識別受體結(jié)合激活小膠質(zhì)細胞,激活的小膠質(zhì)細胞分泌許多炎癥和毒性因子加重神經(jīng)元損傷[17]。而在腦缺血模型中,給予HMGB1中和抗體可抑制缺血后炎癥反應,減輕腦缺血損傷程度,改善腦組織功能[18]。
NMDA組成亞基有三種,NR2亞單位上有與谷氨酸結(jié)合位點,NR2B主要位于前腦和海馬區(qū),在學習和認知功能中發(fā)揮重要作用。在缺血或缺氧、神經(jīng)變性等病理生理情況下,過度激活NR2B可以引起興奮性毒性并導致神經(jīng)元死亡。間歇低氧炎癥可激活iNOS,該酶僅在炎癥情況下表達在小膠質(zhì)細胞上。iNOS可以引起神經(jīng)元去極化和谷氨酸釋放,激活NMDA受體。有研究報道,海馬神經(jīng)元釋放的HMGB1可促進NMDA受體的興奮毒性和功能[6]。本研究發(fā)現(xiàn),胎鼠海馬神經(jīng)元間歇低氧后正常氧培養(yǎng),其HMGB1表達先升高后降低,NMDA表達隨培養(yǎng)時間延長而增加,推測NMDA和HMGB1可能發(fā)揮協(xié)同作用引起神經(jīng)元凋亡和功能障礙。
[1] Lau EY, Choi EW, Lai ES, et al. Working memory impairment and its associated sleep-related respiratory parameters in children with obstructive sleep apnea[J]. Sleep Med, 2015,16(9):1109-1115.
[2] Feng J, Wu Q, Zhang D, et al. Hippocampal impairments are associated with intermittent hypoxia of obstructive sleep apnea[J]. Chin Med J (Engl), 2012,125(4): 696-701.
[3] Gozal E, Gozal D, Pierce WM, et al. Proteomic analysis of CA1 and CA3 regions of rat hippocampus and differential susceptibility to intermittent hypoxia[J]. J Neurochem, 2002,83(2):331-345.
[4] Zhou Q, Sheng M. NMDA receptors in nervous system diseases[J]. Neuropharmacology, 2013,74:69-75.
[5] 明紅,陳銳,王婧,等.海馬神經(jīng)元細胞內(nèi)鈣超載對慢性間歇低氧小鼠認知功能的損傷及鹽酸美金剛的保護作用[J].中華結(jié)核和呼吸雜志,2014,37(12):893-897.
[6] Balosso S, Liu J, Bianchi ME, et al. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons[J]. Antioxid Redox Signal,2014,21(12):1726-1740.
[7] 馮靖,陳寶元,郭美南,等.不同程度、頻率、時限的間歇低氧細胞模型[J].國際呼吸雜志,2007,27(8):578-580.
[8] Ryan S, Taylor CT, McNicholas WT, et al. Selective Activation of Inflammatory Pathways by Intermittent Hypoxia in Obstructive Sleep Apnea Syndrome[J]. Circulation, 2005,112(17):2660-2667.
[9] Wang J, Ming H, Chen R, et al. CIH-induced neurocognitive impairments are associated with hippocampal Ca2+overload, apoptosis, and dephosphorylation of ERK1/2 and CREB that are mediated by overactivation of NMDARs [J]. Brain Res, 2015,1625:64-72.
[10] Yang QW, Xiang J, Zhou Y, et al. Targeting HMGB1/TLR4 signaling as a novel approach to treatment of cerebral ischemia[J]. Front Biosci (Schol Ed), 2010,2:1081-1091.
[11] Zindler E, Zipp F. Neuronal injury in chronic CNS inflammation[J]. Best Pract Res Clin Anaesthesiol, 2010,24(4):551-562.
[12] Kim JB, Sig Choi J, Yu YM, et al. HMGB1, a Novel Cytokine-Like Mediator Linking Acute Neuronal Death and Delayed Neuroinflammation in the Postischemic Brain[J]. J Neurosci, 2006,26(24):6413-6421.
[13] Frank MG, Weber MD, Watkins LR, et al. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming[J]. Brain Behav Immun, 2015,48:1-7.
[14] Kiernan EA, Smith SM, Mitchell GS, et al. Mechanisms of microglial activation in models of inflammation and hypoxia: Implications for chronic intermittent hypoxia[J]. J Physiol, 2016,594(6):1563-1577.
[15] Takata K, Kitamura Y, Tsuchiya D, et al. High mobility group box protein-1 inhibits microglial Aβ clearance and enhances Aβ neurotoxicity[J]. J Neurosci Res, 2004,78(6):880-891.
[16] Laird MD, Shields JS, Sukumari-Ramesh S, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4[J]. Glia, 2014,62(1):26-38.
[17] Gao HM, Zhou H, Zhang F, et al. HMGB1 Acts on Microglia Mac1 to Mediate Chronic Neuroinflammation That Drives Progressive Neurodegeneration[J]. J Neurosci, 2011,31(3):1081-1092.
[18] Liu K, Mori S, Takahashi HK, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats[J]. FASEB J, 2007,21(14):3904-3916.
國家自然科學基金資助項目(81570084)。
馮靖(E-mail: tjzyyfj@163.com)
10.3969/j.issn.1002-266X.2017.02.009
R364.4
A
1002-266X(2017)02-0032-03
2016-09-27)
猜你喜歡
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
娃娃樂園·綜合智能(2020年8期)2020-08-28 00:32:14
創(chuàng)新作文(小學版)(2019年10期)2019-09-25 08:12:28
作文周刊·小學二年級版(2018年9期)2018-04-18 10:01:40
小學生導刊(2018年1期)2018-03-15 08:02:37
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
科技知識動漫(2016年6期)2016-06-24 21:04:53
大灰狼(2015年6期)2015-07-16 21:01:00
作文與考試·小學高年級版(2015年17期)2015-05-30 10:48:04
汽車觀察(2009年1期)2009-02-18 09:11:50
