長QT綜合征的診斷及治療
2016-11-17 21:56:43胡宇才阮燕菲
中西醫結合心血管病電子雜志 2016年17期
關鍵詞:基因突變
胡宇才++阮燕菲

【中圖分類號】R331.3 【文獻標識碼】A 【文章編號】ISSN.2095-6681.2016.17.00.04
1 定義
長QT間期綜合征(LQTS)是以心電圖上QT間期延長為基礎而診斷的,可伴T波及ST段改變,臨床上則表現為易發生惡性室性心律失常如尖端扭轉型室速(Torsades de Pointes,Tdp)、室顫等,患者可有反復發作的黑矇、暈厥,甚至猝死。
2 LQTS的病理生理基礎及病因學分類
2.1 病理生理基礎
各種LQTS共同的病理生理基礎是由于先天性或獲得性原因使心肌細胞膜上的離子通道功能異常,如編碼鈉和鉀離子通道亞單位的基因發生突變導致心肌復極時內向鈉通道失活延遲或鉀離子外流延緩,導致動作電位復極時間延長和復極離散度增大,對應的心電圖變化即為QT間期延長和在此基礎上發生的室性心律失常。
2.2 病因學分類及其表現
2.2.1 遺傳性LQTS 即具有遺傳背景的LQTS,是由于染色體異常或基因突變導致心肌細胞膜上離子通道功能障礙所致,常呈家族性發病。遺傳性LQTS在臨床上又分為兩型:Jervell-Lange-Nielsen(JLN)綜合征和Romano-Ward(RW)綜合征。
2.2.1.1 JLN綜合征
Jervell-Lange-Nielsen(JLN)綜合征為少見類型,是一種伴有神經性耳聾的先天性長QT 間期綜合征,屬于常染色體隱性遺傳病,1957 年首先由Jervell 和Lange、Nielsen 報告,故稱為JLN綜合征,又稱為“聾心綜合征”。臨床上以先天聾啞、QT 間期延長、多形性室性心動過速、尖端扭轉性室性心動過速以及發作性暈厥、心臟性猝死為特征。盡管JLN綜合征為一種罕見疾病,患病率為1.6~6/100萬,但耳聾兒童中本病發病率為0.25~1%。JLN綜合征由編碼心肌細胞鉀通道的KCNQ1或KCNE1基因突變所致,屬常染色體隱性遺傳,父母雙方各帶一個相同或不同的突變并同時把突變傳給子代才會患病,所以病例較少。患兒心電圖上QT間期常大于500ms,伴有雙側嚴重的先天性感覺性聽力喪失。該型患者90%發生過心臟事件,50%在三歲即出現癥狀(暈厥),比其它亞型LQT綜合征患者發病年齡要早,病人首次出現暈厥后10年內病死率接近50%。僅少數JLN綜合征患者對常規β受體阻滯劑和左側心交感神經節切除術(LCSD)治療有效;未經治療的JLN綜合征患者一半以上在15歲前死亡。
2.2.1.2 RW綜合征
Romano-Ward(RW)綜合征占遺傳性LQTS的大部分,是由編碼心肌細胞鉀通道或鈉通道的KCNQ1、KCNH2、KCNE1、KCNE2或SCN5A基因突變所致的一種常染色體顯性遺傳病,心電圖表現為QT間期延長、T波異常以及頻繁的發生尖端扭轉室速(TdP),TdP有自限性,但少數情況下也會蛻變為室顫導致患者猝死。患者聽力正常,可反復發作暈厥,暈厥多在運動或情緒激動時發作,少數在睡眠或休息時發生,發作前常無先兆。攜帶有與RW綜合征相關的突變基因患者中大約50%在一生中會至少有一次暈厥發作,首次發病可以在從嬰兒時期至中年間的任何時候,取決于家族的基因類型,最常見是從十幾歲至二十歲。根據突變基因的不同,RW綜合征可施予β受體阻滯劑或鈉通道阻滯劑治療,左側心交感神經節切除術對部分患者也有效。
2.2.2 獲得性LQTS
即繼發于某些后天性、外源性因素的LQTS,如(1)藥物—包括抗心律失常藥(奎尼丁,索他洛爾,胺碘酮,依布利特)、三環類抗精神病藥、抗微生物藥物(紅霉素,氯奎,金剛胺)、抗組胺藥(阿司咪唑)以及西沙比利、砷劑、有機磷等;(2)電解質紊亂--低鉀,低鈣,低鎂;(3)嚴重緩慢性心律失常;(4)腦血管疾病--腦卒中、腦外傷等;(5)心臟疾病--心肌炎、心肌缺血等;(6)甲狀腺功能減退、低溫、自主神經性疾病等。獲得性LQTS多為可逆性,去除誘因后QT間期可恢復正常。
隨著近年來對LQTS基因型與表型關系研究的深入,發現遺傳性和獲得性LQTS的病例有部分重疊。一些散發的LQTS病例,經基因診斷存在LQTS的遺傳背景但常規心電圖上QT間期并不延長,這些病例僅僅只在有上述獲得性LQTS誘因存在時才表現出QT間期延長及相關的心律失常。
3 遺傳性LQTS的診斷標準
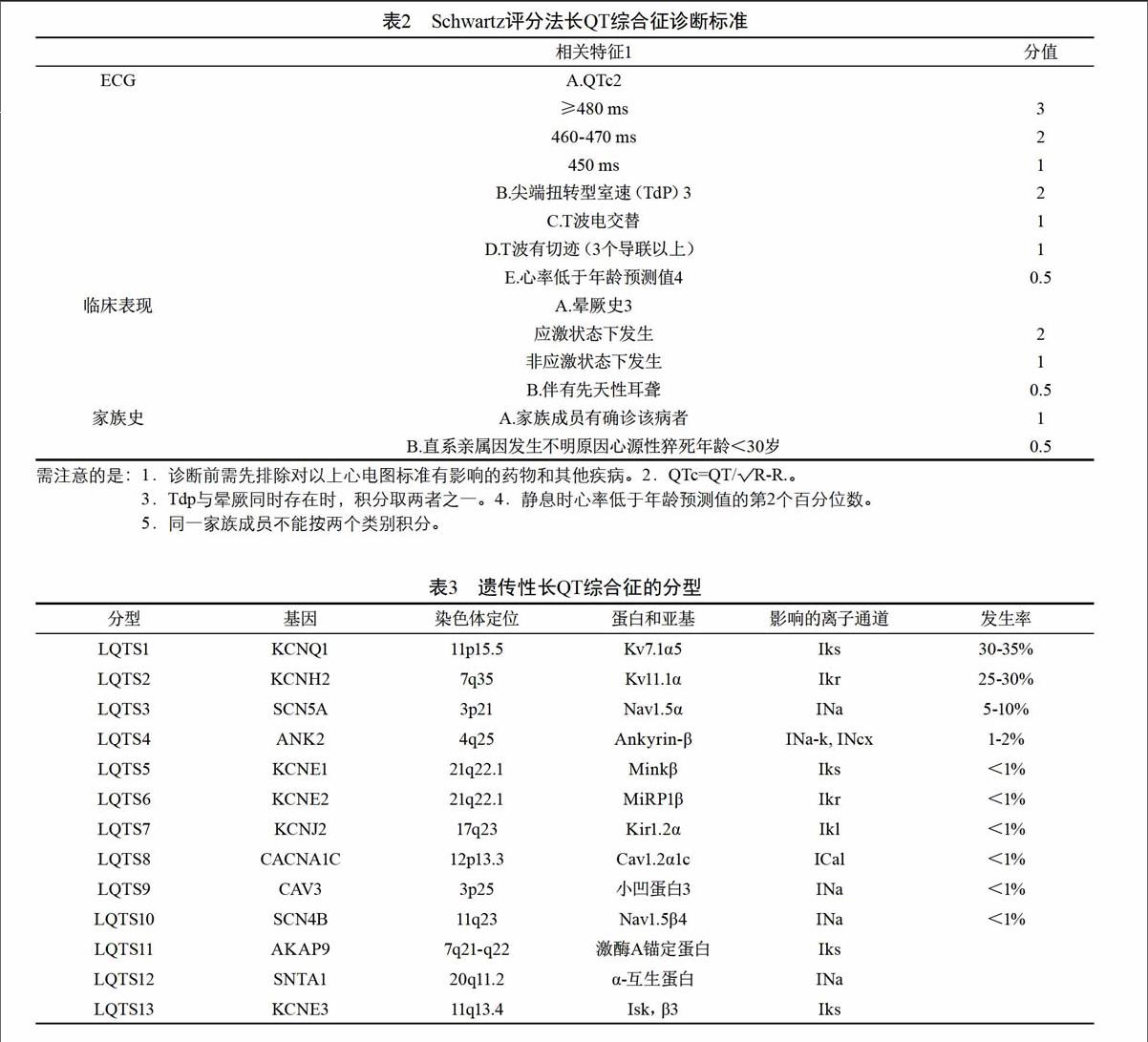
除了排除獲得性LQTS以外,影響QT間期的因素還有很多,如心率、T波終點的確定等,而且所謂“QT間期正常值”也是基于統計學數據的一個區間值(表1),在診斷遺傳性LQTS時還必須個體化。PeterJ.Schwartz于1993年提出的計分法(表2)用于臨床上診斷遺傳性LQTS,敏感性和特異性均較高。按照該計分法,≤1分者,LQTS的診斷可能性小;2~3分者,診斷可疑;≥4分者,診斷肯定。
需注意的是:1.診斷前需先排除對以上心電圖標準有影響的藥物和其他疾病。2.QTc=QT/√R-R.。
3.Tdp與暈厥同時存在時,積分取兩者之一。4.靜息時心率低于年齡預測值的第2個百分位數。
5.同一家族成員不能按兩個類別積分。
4 遺傳性LQTS分型
隨著分子遺傳學研究的進展,很多(75%)遺傳性LQTS的基因突變類型及其相關離子通道功能改變已被闡明,即基因型-表型關系已經明確,并依此對遺傳性LQTS進行了分型(按阿拉伯數字排序),目前已知的有13型-LQTS1、LQTS2、LQTS3…LQTS13(表3)。其中LQTS1、LQTS2和LQTS3占所有已確定基因型遺傳性LQTS病例的95%。
4.1 LQTS1類型特點
LQTS1患者典型的心電圖表現為基底寬大的單向T波,QT間期中度延長,QTc≥500 ms者發生心臟事件的危險性更高。LQTS1患者的心律失常事件大多數由活動誘發,游泳是主要誘因之一。LQTS1對β受體阻滯劑治療反應很好。
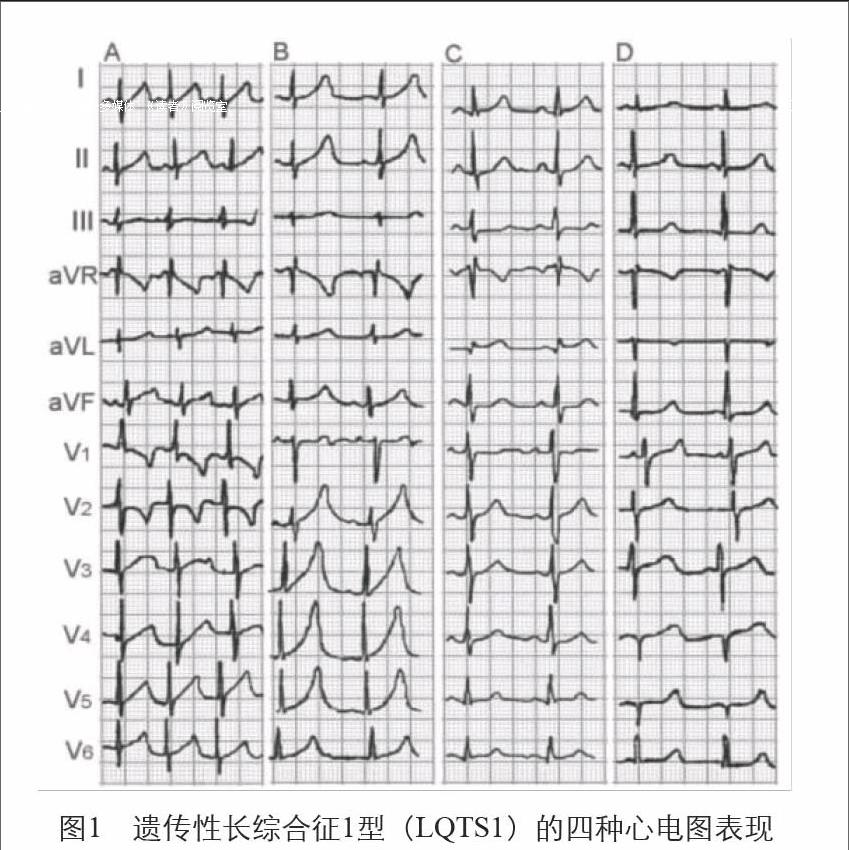
LQT1的心電圖特征:T波基底部增寬,有4種形態:
a.“嬰兒型”T波:T波為非對稱性高聳、基底增寬;b.寬基底T波:T波基底增寬,起始點不明顯;c.T波正常出現,形態正常;d.T波延遲出現,形態正常(圖1)。
LQTS1是遺傳性LQTS的最常見類型,在確定基因型的病例中占50%。緩慢激活延遲整流鉀通道(IKs)的功能缺失是LQTS1的病理生理基礎。1996年,中國學者Wang Q.等首先克隆了位于染色體11p15.5的KCNQ1基因。它由17個外顯子組成,長約400kb,編碼IKs的α亞基。KCNQ1通道是由4個α亞基相互作用形成的四聚體。α亞基含有6個跨膜片段(S1~S6)和一個埋藏在脂質雙層膜中的孔。S4含有許多帶正電荷的氨基酸,是通道的電壓感受器,S5~S6之間的連接區域形成離子的傳導通路。氨基(n-)端和羧基(c-)端位于細胞內。KCNQ1突變有錯義突變、無義突變、剪接突變、氨基酸缺失、移碼突變等,大部分突變發生在跨膜區或細胞內部分。基因突變引起IKs的α亞單位功能受損,IKs減少,動作電位時程延長,心電圖上表現為QT延長。
4.2 LQTS2類型特點
這一類型患者的大多數心律失常事件由情緒激動誘發,鈴聲刺激是LQTS2患者的特異性觸發因素。LQTS2患者對β受體阻滯劑的反應不及LQTS1患者,因此對QT間期顯著延長或反復暈厥的高危患者,可考慮預防性植入ICD并聯合使用β受體阻滯劑。
LQT2的心電圖特征:T波振幅低而有切跡(或雙峰),有6種形態:a.明顯T波雙峰;b.微小的T波雙峰,第二峰出現于T波頂部;c.微小的T波雙峰,第二峰出現于T波降支;d.振幅低平的雙峰T波,第二峰略高;e.振幅較高的雙峰T波,第二峰更高;f.略高于基線的雙峰。(圖2)
LQTS2的致病基因為HERG( human“ether-a-go-go”related gene),位于7q36.1,由4個α亞單位組成,每個亞單位有6個跨膜片段,1個孔區及氨基和羧基末端,n和c末端均位于細胞內,跨膜片段s5、s6及連接兩者的胞內肽段共同構成通道孔的結構域,跨膜區與孔區是突變的好發位點。HERG編碼快速激活的延遲整流鉀通道(IKr)的α亞基,IKr是心室肌細胞動作電位復極期中的主要外向電流。HERG基因突變則導致IKr通道失活,外向鉀電流減少或消失,引起心肌復極時間(QT間期)延長。HERG突變涉及通道的各個區域,目前將HERG突變致LQTS2的機制歸納為:①合成異常;②細胞內運輸異常;③通道門控異常;④通道離子選擇性異常。一種突變可通過4種機制中的一種或數種發揮作用,其中合成異常和細胞內運輸異常是HERG基因錯義突變導致LQTS2的最常見機制。
4.3 LQTS3類型特點
LQTS3的患病數在所有確定基因型的LQTS患者中約占10-15%。LQTS3的典型心電圖表現為ST段延長和小而尖的T波,大多數心律失常事件發生于休息狀態或睡眠中。患者對β受體阻滯劑反應差,部分患者使用鈉通道阻滯劑可減少暈厥或TdP發作,高危的LQTS3患者應植入ICD。
LQT3的心電圖特征:ST段延長,T波延遲出現,嬰幼兒期易發生2:1房室阻滯,T波形態有2種:a. T波延遲出現,高聳或呈雙相;b. T波非對稱性高聳。須注意的是在同一家系中,長QT綜合征患者之間,T波形態可有重疊;而不同致病基因之間T波形態也有重疊。(圖3)
LQTS3的致病基因是編碼心臟門控鈉通道的SCN5A,定位于3p21,由28個外顯子組成,編碼一由2016個氨基酸組成的蛋白質。該蛋白在細胞膜上形成4個結構類似的同源結構域,每個區域由6個跨膜片段組成,形成鈉通道α單位;其中s5和s6片段之間的連接環構成通道孔,通道孔具有不對稱結構,選擇性通過Na離子。s4片段為通道的電壓感受器,當細胞膜電位除極時可使s4片段發生跨膜移動,激活鈉通道產生鈉電流。SCN5A基因突變可改變鈉通道的正常結構,進而改變鈉通道的功能,并導致心律失常的發生。突變使鈉通道失活延遲,2相的Na電流持續不失活,使復極和動作電位的時程延長。由于動作電位時程的異常延長,使早期后除極及觸發活動增加,并易誘發尖端扭轉型室速。研究表明鈉通道的失活延遲與心率減慢有關,因而LQTS3患者多在心率緩慢或睡眠時發生心律失常事件。
5 LQTS心臟以外的表現
目前發現LQTS7與LQTS8患者有心臟以外的臨床表現,有助于鑒別診斷。
5.1 Andersen-Tawil綜合征
即LQTS7,是一種常染色體顯性遺傳病,其特征為周期性麻痹、QT間期延長伴心律失常、機體畸形三聯征,其中典型的畸形包括寬眼距、小下頜、低耳廓、第5指(趾)彎曲、第2和3指(趾)并指(趾)、身材矮小以及脊柱側凸等。70%的LQTS7與KCNJ2基因突變有關,后者使內向整流鉀電流(KIr2.1)減小,引起延遲后除極和心律失常,心電圖上可表現為QT間期輕度延長、寬大U波等。Andersen-Tawil綜合征患者在幼年或少年時期出現心臟癥狀(心悸、暈厥)和/或運動障礙。
5.2 Timothy綜合征
即LQTS8,是編碼電壓門控的鈣離子通道基因(CACNA1C)的突變所致的一種較罕見的LQTS臨床表型。由于離子通道失活鈍化,導致鈣超載從而引起外周組織發育和功能異常,同時伴有心電圖改變和心律失常。心電圖上可見顯著延長的QT間期、房室傳導阻滯和巨大的T波。心外表現中最突出的是并趾畸形,一部分Timothy綜合征兒童還可有免疫缺陷、認知障礙、間隙性低血糖以及孤獨癥。Timothy綜合征惡性程度較高,大部分患兒死亡時的平均年齡為2.5歲。
6 LQTS的治療原則
獲得性LQTS通常不需要長期治療,去除引起QT間期延長的原因后,QT間期多可恢復正常。
遺傳性LQTS患者必須進行長期預防性的治療以降低猝死的風險。β受體阻滯劑能顯著減少心律失常事件的發生,在LQTS1患者療效更為明顯,對于有明顯心動過緩的患者,可在安裝了永久起搏器后維持β受體阻滯劑治療。不能耐受β受體阻滯劑或治療效果不佳的患者,左側心交感神經節切除術(LCSD)可作為二線治療。對于接受足量β受體阻滯劑和LCSD治療或安裝了永久起搏器后仍反復發生暈厥/Tdp的
患者應考慮植入ICD,ICD雖不能預防惡性心律失常的發生,但可有效預防猝死。
由于遺傳性LQTS的治療和預后有著鮮明的基因特異性,基因學結果指導下的個體化治療策略將在很大程度上提高LQTS患者及其家族成員的治療效果。盡管目前進行基因測定和分型的價格還很昂貴,但隨著分子生物學的發展和商業運作的參與,遺傳性LQTS的基因診斷和治療必定成為一種趨勢而得到廣泛推廣應用。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
