HPLC梯度洗脫法測定對乙酰氨基酚口服混懸液中有關物質
2016-11-04 05:44:05余俊玲徐偉斌
北方藥學 2016年10期
關鍵詞:檢測
余俊玲 鐘 瑜 徐偉斌
(廣州博濟醫藥生物技術股份有限公司廣州510700)
HPLC梯度洗脫法測定對乙酰氨基酚口服混懸液中有關物質
余俊玲鐘瑜徐偉斌
(廣州博濟醫藥生物技術股份有限公司廣州510700)
目的:采用高效液相色譜法(HPLC)梯度洗脫測定對乙酰氨基酚口服混懸液中有關物質含量。方法:用Inertsustain C18(250mm×4.6mm,5μm)為色譜柱;以0.05mol·L-1醋酸銨溶液和甲醇為流動相進行梯度洗脫;檢測波長為245nm,流速為1.0mL·min-1,柱溫為25℃。結果:各峰間能完全分離;對乙酰氨基酚和對氨基酚的最低檢測濃度分別為0.027μg·mL-1和0.093μg·mL-1;對乙酰氨基酚在0.037~0.33μg·mL-1范圍內峰面積與濃度呈良好的線性關系,對氨基酚在0.26~3.09μg·mL-1范圍內峰面積與濃度呈良好的線性關系;中間精密度試驗對氨基酚RSD為13.77%。結論:該方法專屬性好、靈敏、結果準確,可用于對乙酰氨基酚口服混懸液的有關物質含量控制。
高效液相色譜法 梯度洗脫 對乙酰氨基酚口服混懸液 對氨基酚 有關物質
對乙酰氨基酚是目前主要用于解熱鎮痛的藥物,抗炎作用極弱,對胃腸道無明顯刺激,為止痛首選藥物之一。藥品的質量、安全均與有關物質有關。對氨基酚是對乙酰氨基酚生產時帶入或對乙酰氨基酚及其制劑儲存時產生的降解產物,對人體有一定的毒性[1],因此要嚴格控制對氨基酚及其他有關物質。2015版《中國藥典》[2]收載有對乙酰氨基酚原料及制劑的質量標準,有關物質的檢測采用了HPLC法。有文獻報道[3]測定對乙酰氨基酚口服溶液中的有關物質,采用HPLC等度洗脫法,但戴正琳[3]最后在文中提出采用HPLC梯度洗脫法更優。也有文獻報道[4]采用HPLC梯度洗脫法測定對乙酰氨基酚原料中有關物質,未見采用HPLC梯度洗脫法測定口服混懸液中的有關物質報道。本文采用HPLC梯度洗脫法測定對乙酰氨基酚口服混懸液中的有關物質,方法專屬、靈敏、結果準確,可用于對乙酰氨基酚口服混懸液的質量控制。
1 儀器與試藥
e2695型高效液相色譜儀(美國沃特世公司,DAD,二極管陣列檢測器),LC-20AT型高效液相色譜儀(日本島津公司),CPA 225D型電子分析天平(德國賽多利斯公司);對乙酰氨基酚對照品來自中國食品藥品檢定研究院,批號100018-201409,對氨基酚對照品來自中國食品藥品檢定研究院,批號00802-201203;對乙酰氨基酚口服混懸液(泰諾林,上海強生制藥有限公司,批號:150426216);甲醇為色譜純,醋酸、醋酸銨為分析純,水:屈臣氏蒸餾水。
2 方法與結果
2.1色譜條件:色譜柱:島津Inertsustain C18鍵合硅膠柱(250mm×4.6mm,5μm);以0.05mol·L-1醋酸銨溶液為流動相A,以甲醇為流動相B按表1進行梯度洗脫,檢測波長為245nm,流速為1.0mL·min-1,柱溫為25℃,進樣量為10μL;稀釋劑為0.05mol·L-1醋酸銨溶液-甲醇(85∶15)。
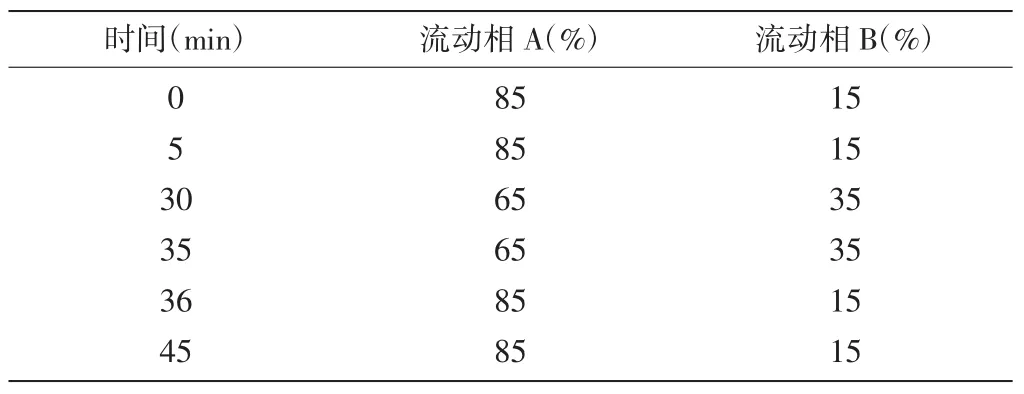
表1 梯度洗脫表
2.2系統適用性:精密稱取本品、對氨基酚對照品適量加稀釋劑溶解并稀釋制成每1mL約含2mg對乙酰氨基酚和每1mL約含4μg對氨基酚的混合溶液;再精密稱取陰性樣品、原料適量分別加稀釋劑溶解并稀釋制成每1mL相當含6mg對乙酰氨基酚的陰性溶液和每1mL約含2mg對乙酰氨基酚的原料供試液,取各供試液濾過,按“2.1”項下色譜條件檢測,記錄色譜圖,以系統適用性色譜圖計,理論塔板數按對乙酰氨基酚峰計為9522,對乙酰氨基酚與對氨基酚分離度為18,與其他各峰分離度均大于1.5(見圖1)。
2.3專屬性
2.3.1未破壞試驗:精密稱取本品適量,用稀釋劑配制成每1mL約含2.5 mg的對乙酰氨基酚溶液,濾過,按“2.1”項下色譜條件檢測,記錄色譜圖(見圖2A)。
2.3.2氧化破壞試驗:精密稱取本品適量,加入30%H2O2溶液1mL,常溫放置1h,用稀釋劑配制成每1mL約含2.5mg的對乙酰氨基酚溶液,濾過,按“2.1”項下色譜條件檢測,記錄色譜圖(見圖2B)。
2.3.3酸破壞試驗:精密稱取本品適量,加入1mL 1mol·L-1的鹽酸溶液,置60℃水浴2.5h,待冷卻至室溫,用1mol·L-1氫氧化鈉溶液調pH至中性,用稀釋劑配制成每1mL約含2.5mg的對乙酰氨基酚溶液,濾過,按“2.1”項下色譜條件檢測,記錄色譜圖(見圖2C)。
2.3.4堿破壞試驗:精密稱取本品適量,加入1mL 1mol·L-1的氫氧化鈉溶液,置60℃水浴2.5h,待冷卻至室溫,用1mol·L-1鹽酸溶液調pH至中性,用稀釋劑配制成每1mL約含2.5mg的對乙酰氨基酚溶液,濾過,按“2.1”項下色譜條件檢測,記錄色譜圖(見圖2D)。
2.3.5高溫破壞試驗:精密稱取本品適量,置60℃水浴6h,待冷卻至室溫,用稀釋劑配制成每1mL約含2.5 mg的對乙酰氨基酚溶液,濾過,按“2.1”項下色譜條件檢測,記錄色譜圖(見圖2E)。
2.3.6強光破壞試驗:精密稱取本品適量,置紫外燈下照射6h,用稀釋劑配制成每1mL約含2.5mg的對乙酰氨基酚溶液,濾過,按“2.1”項下色譜條件檢測,記錄色譜圖(見圖2F)。
綜上所述,各條件下,主峰與其他峰、其他峰之間均能達到基線分離,說明方法專屬性強,可用于本品的雜質檢測。
2.4檢測限和定量限:精密稱取對乙酰氨基酚、對氨基酚對照品適量,用稀釋劑溶解并稀釋制成每1mL約含2.5mg的對乙酰氨基酚溶液和每1mL約含2.6μg的對氨基酚溶液,再分別稀釋數倍制成系列溶液,按“2.1”項下色譜條件檢測,當信噪比(S/N)約為3時,檢測限分別為0.027μg·mL-1和0.093μg·mL-1;當信噪比(S/N)約為10時,定量限分別為0.041μg·mL-1和0.22μg·mL-1。
2.5精密度試驗
2.5.1進樣精密度:取“2.2”項下系統適用性溶液,濾過,按“2.1”項下色譜條件連續進樣6次,記錄色譜圖,以峰面積計算對氨基酚RSD為1.29%,其他雜質峰RSD分別為1.62%、0.97%、0.58%。結果表明進樣精密度良好。
2.5.2中間精密度:精密稱取本品、對氨基酚對照品適量加稀釋劑溶解并稀釋制成每1mL約含2.5mg的對乙酰氨基酚和每1mL約含2.6μg對氨基酚混合溶液6份,濾過,作為供試品溶液;精密量取1mL置100mL量瓶中,加稀釋劑稀釋至刻度,作為對照溶液;另精密稱取對氨基酚對照品適量,加稀釋劑溶解并稀釋制成每1mL約含2.6μg的溶液,作為對照品溶液。分別由不同人員在不同儀器按“2.1”項下色譜條件檢測,計算對氨基酚含量及其他各雜質量。對氨基酚平均含量為0.034%,RSD為13.77%,未知雜質1平均為0.008%,RSD為20.31%,未知雜質2平均為0.041%,RSD為15.79%,未知雜質3平均為0.037%,RSD為16.23%。結果表明,中間精密度均符合要求。
2.6線性范圍:精密稱取對乙酰氨基酚、對氨基酚對照品適量,用稀釋劑溶解并稀釋制成0.037~0.33μg·mL-1和0.26~3.09μg· mL-1的系列濃度溶液,按“2.1”項下色譜條件檢測,記錄色譜圖,分別以峰面積(A)對濃度(C)進行線性回歸,結果回歸方程分別為:A=37728C-0.220,r=0.998;A=14215C+351.9,r=0.998。
2.7回收率:精密稱取對氨基酚對照品適量用稀釋劑溶解并配制成每1mL約含20.0μg的對氨基酚溶液,精密移取3mL置25mL量瓶中,用稀釋劑稀釋至刻度,搖勻,作為對照品溶液;精密稱取本品適量(約相當于對乙酰氨基酚125mg),分別置11個50mL量瓶中,分別加適量稀釋劑溶解,取9個分組,每3個為1組,每組分別加入對氨基酚溶液4mL、6mL、8mL,用稀釋劑稀釋至刻度,搖勻,作為供試品溶液,另2個分別用稀釋劑稀釋至刻度,搖勻,作為陰性供試品溶液;取對照品溶液和各供試品溶液,濾過,按“2.1”項下色譜條件檢測,計算回收率,試驗結果見表2,平均回收率為98.3%,RSD為2.91%,該方法準確度良好。
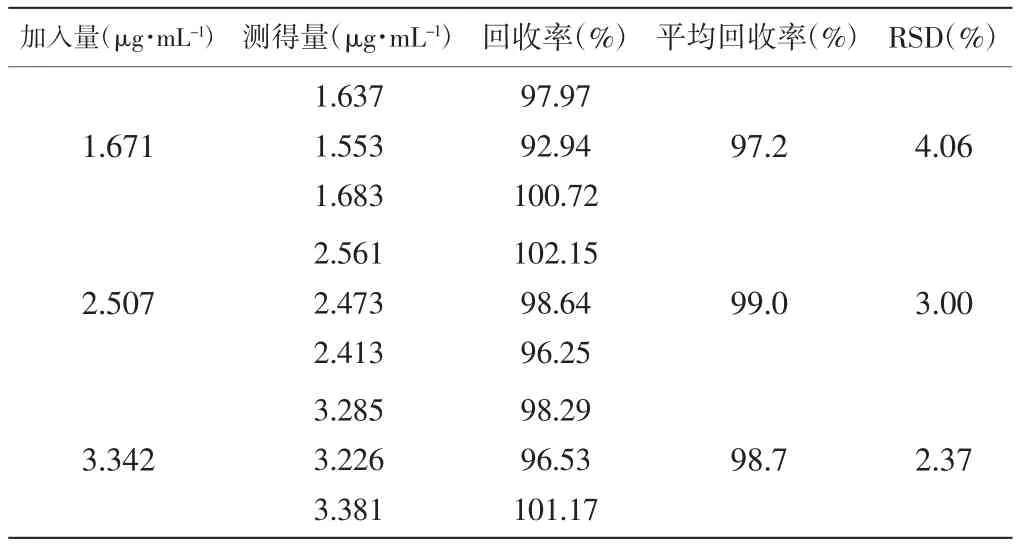
表2 回收率試驗結果
2.8溶液穩定性
2.8.1對氨基酚溶液穩定性:精密稱取對氨基酚對照品適量,用稀釋劑溶解并稀釋制成2.6μg·mL-1的溶液,分別在0、2、4、6h按“2.1”項下色譜條件測定,記錄色譜圖,以峰面積計算RSD為1.77%。結果表明該溶液在6h內基本穩定。
2.8.2加對氨基酚的樣品溶液穩定性:取“2.5.2”項下供試品溶液1份,分別在0、2、4、6、8h按“2.1”項下色譜條件測定,記錄色譜圖,以峰面積計算對氨基酚RSD為1.63%,其他雜質峰RSD分別為1.58%、0.89%、0.76%。結果表明該溶液在8h內基本穩定。2.9耐用性試驗:微小調整“2.1”項下的流動相比例、流速、柱溫及色譜柱后,取“2.2”項下系統適用性溶液進行試驗,記錄色譜圖,用分離度進行評價。結果表明,選定的色譜條件進行微小調整后,各峰之間分離度均符合要求,耐用性良好。
3 方法的比較
取本品適量,加稀釋劑溶解稀釋制成溶液,濾過,按“2.1”項下色譜條件測定,并與現行方法[5]進行比較,擬定方法較現行方法檢測靈敏度更高,雜質檢出數更多,具體結果見表3。
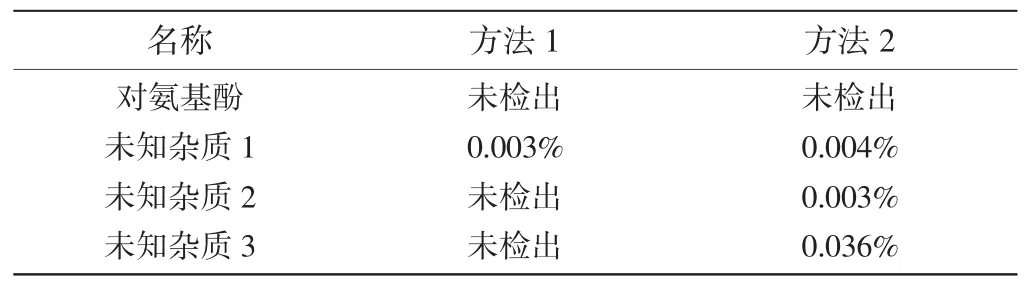
表3 方法比較結果
4 討論
4.1取本品在氧化、酸、堿、高溫、強光條件破壞后,在擬定色譜條件下檢測,各峰間均能達到基線分離,各峰純度均符合要求,各條件下物料平衡。
4.2溶液穩定性的考察結果表明,供試品溶液及對氨基酚溶液均應臨用新制。
4.3因《中國藥典》[2]規定對氨基酚不得過對乙酰氨基酚標示量的0.1%,其他各雜質和不得大于1.0%,制定一個方便的測定雜質方法很重要,本文擬定方法測定雜質操作簡單,專屬性強,結果準確可靠。
[1]莊幼齡,邱麒.HPLC法測定對乙酰氨基酚緩釋干混懸劑的有關物質[J].海峽藥學,2010,22(2):45-48.
[2]中國藥典.2015.Vol(二部):318-323.
[3]戴正琳,陶志.HPLC法測定對乙酰氨基酚口服溶液中的有關物質對氨基酚[J].藥物分析雜質,2011,31(4):785-787.
[4]趙金會,王金彬.HPLC梯度洗脫法測定對乙酰氨基酚原料藥及有關物質對氨基酚[J].北方藥學,2012,9(10):1-2.
[5]新藥轉正標準[S].第77冊,149.
Determinarion of related substances in paracetamol oral suspension using HPLC with gradient elution
Yu Junling Zhong yu Xu Weibin(GuangZhou boji Medical Biotechnological Co.Ltd,Guangzhou 510700,China)
Objective:To established an HPLC method for the determination of related substances in paracetamol oral suspension.Methods:A Inertsustain C18column(250mm×4.6mm,5μm)was adopted.The mobile phase of gradient elution was ammonium acetate solution of 0.05mol·L-1and methanol.The wavelength of detector was 245nm,the flow rate was 1.0mL·min-1,the Column temperature was 25℃.Results:Any two peaks was completely separated.The detected limit were 0.027μg·mL-1and 0.093μg·mL-1for paracetamol and 4-aminophenol.The calibration curve was l inear in the range of 0.037~0.33μg·mL-1and 0.26~3.09μg·mL-1.the RSD was 13.77%(n=12)for 4-aminophenol.Conclusion:The proposed HPLC method could be used for the determination of reltated substances in paracetamol oral suspension.
HPLC Gradient elution Paracetamol oral suspension 4-aminophenol Related matt
R927.2
A
1672-8351(2016)10-0005-03
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
