氟代苯基異羥肟酸鈷的合成及其對乙苯氧化的催化性能
2016-10-25 03:00:33陳小峰呂新宇
合成化學 2016年9期
關鍵詞:催化劑
陳小峰, 邱 滔, 呂新宇
(常州大學 設計研究院,江蘇 常州 213164)
?
氟代苯基異羥肟酸鈷的合成及其對乙苯氧化的催化性能
陳小峰, 邱滔*, 呂新宇
(常州大學 設計研究院,江蘇 常州213164)
以全氟碘丁烷為原料,分別與4-碘硝基苯和4-碘苯甲酸反應合成了4-全氟丁基硝基苯(1)和4-全氟丁基苯甲酸(3); 1經還原反應, 3經酰氯化反應,后再縮合反應制得氟代苯基異羥肟酸(5); 5經絡合反應合成了氟代苯基異羥肟酸鈷(6), 其結構經 UV-Vis,1H NMR, FT-IR和HR-MS(EI)表征。在氟兩相中考察了其對乙苯氧化的催化性能。結果表明:在全氟己烷中,6 0.04 mmol,于60 ℃反應6 h,乙苯的轉化率為49.2%,苯乙酮的選擇性為88.3%。6循環使用5次,選擇性保持良好。
氟代苯基異羥肟酸鈷; 氟兩相催化; 乙苯氧化; 合成; 催化性能
氟兩相催化作為一種新型均相催化和分離技術,在1994年Horvath首次使用后便逐漸成為研究熱點[1]。氟兩相體系由氟相和有機相構成,氟相由全氟溶劑和氟代催化劑構成,氟兩相具有升溫后有機相和氟相形成均相體系而降溫后兩相分離的特性,故適合用于催化體系[2-3]。開發合適的氟代催化劑是氟兩相催化的關鍵[4-6]。異羥肟酸結構簡單,合成簡便,結構可調性大,可作為一種新的金屬有機催化劑載體[7-10]。研究表明:在異羥肟酸配體結構上引入基團能增加赤道配體的π-授予性、疏水性,能提高配合物結合氧的能力,從而提高催化氧化性能[11-12]。

Scheme 1
異羥肟酸金屬配合物和氟兩相體系結合作為催化氧化的報道較少。本文以全氟碘丁烷為原料,分別與4-碘硝基苯和4-碘苯甲酸反應合成了4-全氟丁基硝基苯(1)和4-全氟丁基苯甲酸(3); 1經還原反應,3經酰氯化反應,后經縮合反應制得氟代苯基異羥肟酸(5); 5經絡合反應合成氟代苯基異羥肟酸鈷(6),其結構經 UV-Vis,1H NMR, FT-IR和HR-MS(EI)表征。在氟兩相中考察了其對乙苯氧化的催化性能。
1 實驗部分
1.1儀器與試劑
Nicolet 380型傅立葉變換紅外光譜儀(KBr壓片); Avance 500 MHz型超導傅立葉變換核磁共振儀(CDCl3為溶劑,TMS為內標);Ionspec 4.7 Tesla FTMS型高分辨質譜儀;N2000型氣相色譜儀;LC-100型液相色譜儀;QP2010 SE型氣相色譜質譜聯用儀。
全氟碘丁烷、全氟己烷(質量分數≥99.5 %),百靈威科技有限公司;其余所用試劑均為分析純。
1.2合成
(1) 1的合成
在反應瓶中依次加入活性銅0.77 g(12 mmol),全氟碘丁烷4.15 g(12 mmol) 和DMSO 4 mL,攪拌下于80 ℃反應0.5 h;加入4-碘硝基苯 1.00 g(4 mmol),反應至終點(TLC檢測)。加冰水75 mL,用乙酸乙酯萃取,萃取液蒸干后經硅膠柱層析[洗脫劑:A=V(乙酸乙酯) ∶V(石油醚)=1 ∶7]純化得黃色固體1 0.89 g,收率65%;1H NMRδ: 8.35(d,J=8.92 Hz, 2H), 7.82(d,J=8.72 Hz, 2H);19F NMRδ: -76.79~-77.30(m, 3F, CF3), -115.06~-118.50(m, 2F, CF2), -128.39~-128.49(m, 2F, CF2), -134.57~-134.96(m, 2F, CF2); GC-MSm/z: 340{[M-H]-}。
(2) 4-全氟丁基苯基羥胺(2)的合成
在反應瓶中加入1 2.00 g(5.8 mmol)和無水乙醇20 mL,攪拌下分批加入鋅粉1.00 g(15 mmol),甲酸銨0.60 g(9.5 mmol)和水2 mL,加畢(30 min),反應至終點(TLC檢測)。用水淬滅反應,用乙酸乙酯萃取,萃取液旋蒸除溶得黃色晶體2 1.36 g,收率72%;1H NMRδ: 7.55(d,J=8.72 Hz, 2H), 6.77(d,J=8.92 Hz, 2H);19F NMRδ: -76.79~-77.30(m, 3F, CF3), -115.06~-118.50(m, 2F, CF2), -128.39~-128.49(m, 2F, CF2), -134.57~-134.96(m, 2F, CF2); LC-MSm/z: 326.02{[M-H]-}。
(3) 3的合成
在反應瓶中加入活性銅2.00 g(0.03 mol),全氟碘丁烷10.38 g(0.03 mol)和DMSO 4 mL,于80 ℃靜置反應0.5 h,加入4-碘苯甲酸1.00 g(4 mmol),攪拌下反應至終點(TLC檢測)。加冰水75 mL,過濾,用乙酸乙酯萃取,萃取液蒸除溶劑得固體粗產物。固體粗產品經堿液溶解,鹽酸酸化得淡黃色固體3 0.96 g,收率71%; LC-MSm/z: 339{[M-H]-}。
(4) 4-全氟丁基苯甲酰氯(4)的合成
在反應瓶中加入3 0.80 g(1.8 mmol),氯化亞砜8 mL和DMF 1滴,攪拌下于80 ℃反應3 h。蒸除氯化亞砜得黃色油狀液體4 0.63 g,收率98%, LC-MSm/z: 357{[M-H]-}。
(5) 5的合成
在反應瓶中加入2 1 .00 g(3 mmol),二氯甲烷30 mL和NaHCO30.18 g,攪拌下于-10~-5 ℃滴加4 1.07 g(3 mmol)的二氯甲烷(20 mL)溶液,滴畢(30 min),于室溫反應2~4 h(TLC檢測)。過濾,濾餅經柱層析(洗脫劑:A)純化得黃色晶體5 0.82 g,收率55%;1H NMRδ: 7.82(d,J=8.92 Hz, 2H), 7.73(d,J=8.72 Hz, 2H), 7.49(d,J=8.92 Hz, 2H), 7.44(d,J=8.72 Hz, 2H);19F NMR(470 Hz)δ:-80.949~-80.991(m, 3F, CF3), -111.473~-111.529(m, 2F, CF2), -122.627~-122.667(m, 2F, CF2), -125.515~-125.569(m, 2F, CF2); IRν: 3 188(OH), 1 636(C=O), 1 353(CF3), 1 237, 1 205, 1 135, 1 090(CF2), 1 009(C—F), 908, 745, 735; LC-MSm/z: 648.01{[M-H]-}。
(6) 6的合成
在反應瓶中加入5 1 mmol和無水乙醇10 mL,攪拌下于50 ℃滴加乙酸鈷水溶液2 mL,滴畢,反應3 h。于沸水浴加熱30 min,過濾,濾餅用熱水和95%乙醇洗滌,于100 ℃真空干燥3~4 h得粉紅色固體6 0.73 g,收率53%, ICP測試鈷含量為3.39%(理論含量為3.3%); IRν: 1 630(C=O), 1 353(CF3), 1 237, 1 205, 1 135, 1 090(CF2), 1 009(C—F), 908, 745, 735; HR-MS(EI)m/z: Calcd for C42H4N2O4F36Co {[M+Na]+}1 355.063, found 1 378.052。
1.3氟兩相催化氧化乙苯反應
在反應瓶中依次加入6 0.072 g (0.041 mmol), C6F146 mL (29.6 mmol)和乙苯10 mL (82 mmol),以空氣為氧源,于60 ℃持續通入空氣反應6 h。冷卻至室溫,有機相與氟碳溶劑分層,取上層有機相分析產物及含量(氣相色譜初始溫度50 ℃,終止溫度250 ℃,程序升溫20 ℃·min-1)。
2 結果與討論
2.1表征

λ/nm
圖1是6的UV-Vis 譜圖。由圖1可見,在300 nm和550 nm 處有吸收峰,表明其形成了配合物結構[10]。結合HR-MS(EI)譜圖,可以說明所得產物為目標化合物。
2.2催化性能研究
以催化氧化乙苯(Scheme 2)為模板反應,考察6在氟兩相催化中的性能。分別考察了催化劑用量,反應時間,反應溫度和回收次數對乙苯氧化的影響。

Scheme 2
(1) 催化劑用量
以6為催化劑,于60 ℃反應6 h,考察了不同催化劑用量對乙苯催化氧化反應的影響。其結果見表1。
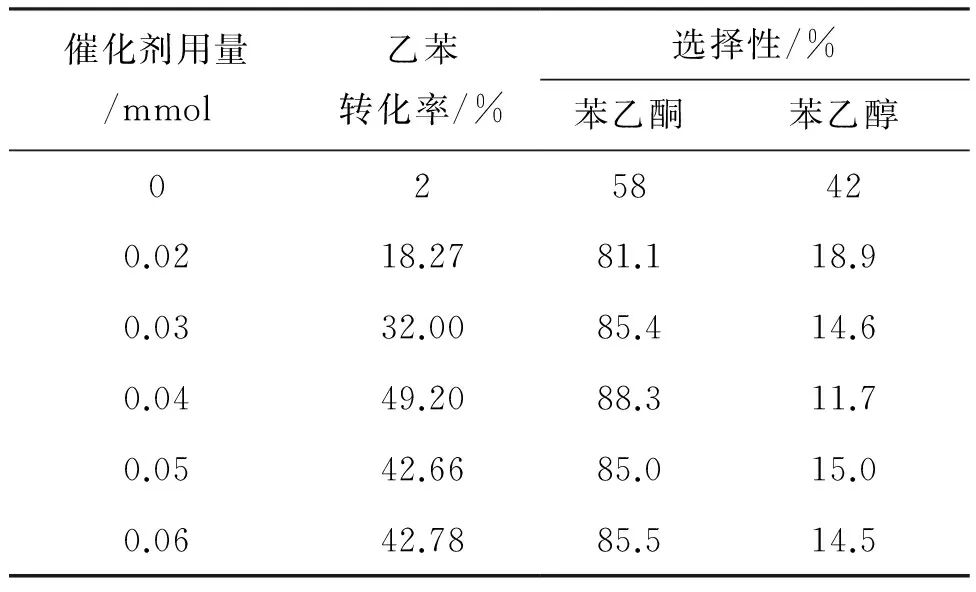
表1 6用量對乙苯氧化的影響
由表1可見,催化劑用量對乙苯轉化率影響較大,對苯乙酮選擇性影響不大。未加入催化劑時,乙苯的轉化率很低;當催化劑用量為0.04 mmol時,乙苯的轉化率最大(49.20%)。當催化劑的量進一步加大時,催化劑分子之間會聚集[13],使得活性組分的有效活性面積減小,不利于其與氧氣的結合,從而導致催化效率下降。
(2) 反應時間
6 0.04 mmol,反應溫度60 ℃,考察了反應時間對乙苯氧化反應的影響,結果見表2。
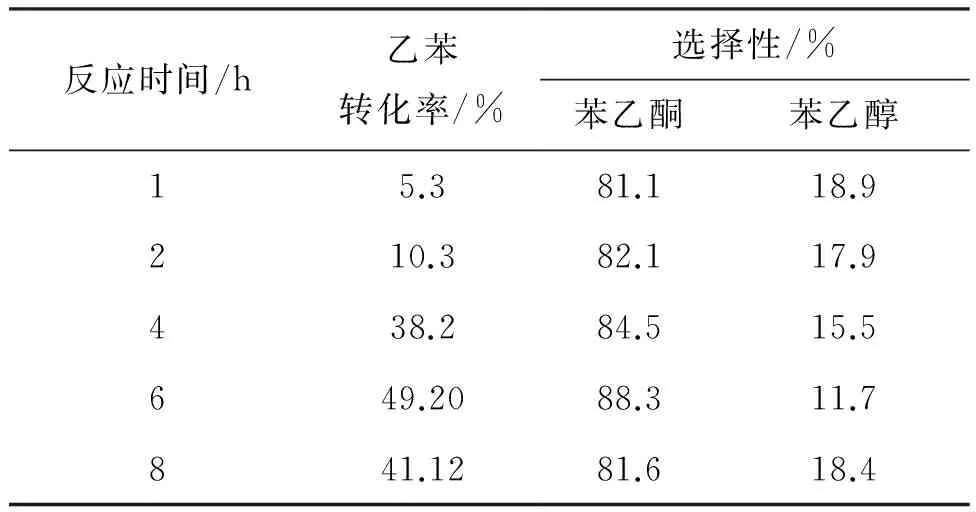
表2 反應時間對乙苯氧化的影響
由表2可見,隨著反應時間增加,乙苯的轉化率增大,氧化反應速度明顯加快,苯乙酮的選擇性變化不大,說明該反應具有明顯的誘導期。 反應時間為6 h 時,轉化率趨于平緩,同時轉化率和選擇性都達到最大值,分別為49.20%和88.3%。隨著時間的延長,選擇性降低明顯,這是由于苯乙酮發生深度氧化[14]。
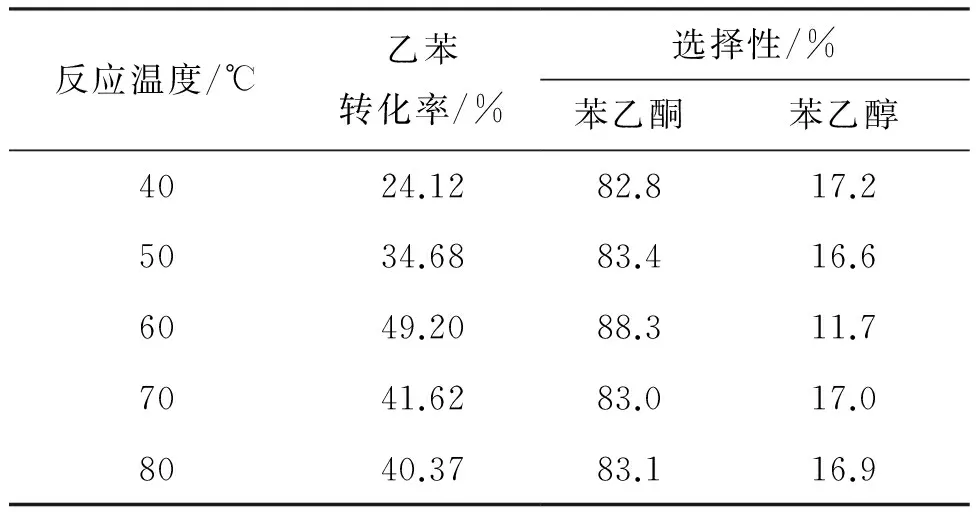
表3 溫度對乙苯氧化的影響
(3) 反應溫度
6 0.04 mmol,反應時間為6 h,考察溫度對乙苯氧化反應的影響,結果見表3。由表3可見,隨著溫度的升高,乙苯轉化率和選擇性均有所增加,且反應速率也逐漸提高,但溫度達到60 ℃以后,不僅轉化率下降,其選擇性也下降。故最佳反應溫度為60 ℃。導致這種現象可能有兩種原因:第一,氧氣溶解度下降;第二,升高溫度也加速了氧化產物進一步被氧化[15]。
(4) 回收次數對乙苯氧化的影響
根據氟碳溶劑的熱力學特性分離回收全氟溶劑及氟代催化劑,再應用于乙苯的催化氧化實驗中,回用7次得出的結果見表4。
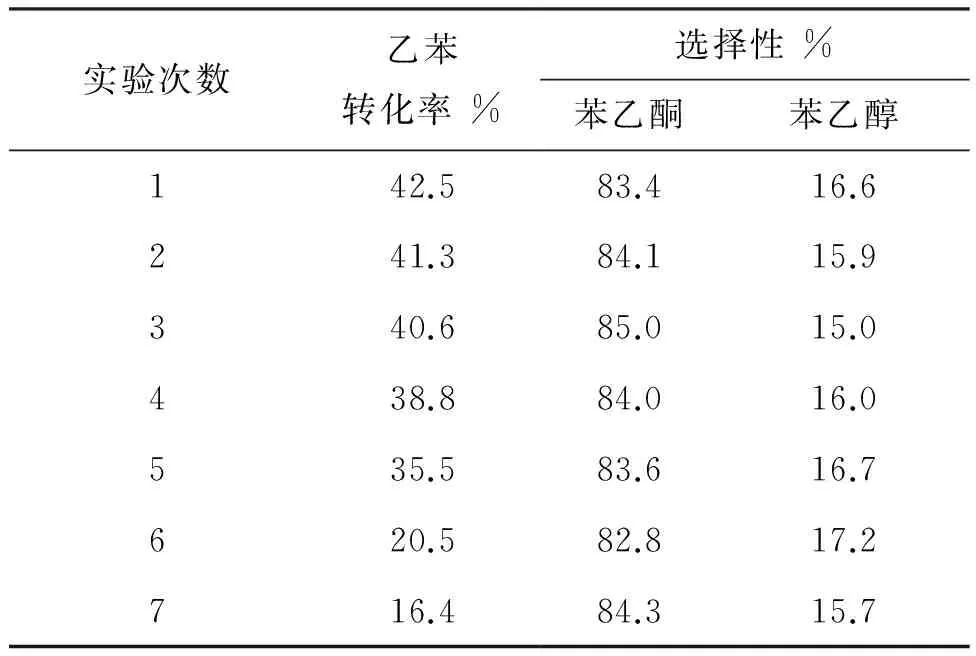
表4 回收次數對乙苯氧化的影響
由表4可以看出,氟碳相重復使用5次后乙苯轉化率下降明顯,但苯乙酮的選擇性保持良好,由此說明6在多次使用后選擇性依舊保持良好。
以全氟碘丁烷為原料,分別與4-碘硝基苯和4-碘苯甲酸反應合成了4-全氟丁基硝基苯(1)和4-全氟丁基苯甲酸(3); 1經還原反應,3經酰氯化反應,后經縮合反應制得氟代苯基異羥肟酸(5); 5經絡合反應合成氟代苯基異羥肟酸鈷(6)。在氟兩相中考察了其對乙苯氧化的催化性能。結果表明:在全氟己烷中, 6 0.04 mmol,于60 ℃反應6 h,乙苯的轉化率為49.2%,苯乙酮的選擇性為88.3%,催化劑循環使用5次,催化劑選擇性保持良好。6在氟兩相催化中的應用提供了一種綠色環保的催化氧化工藝,對拓展氟兩相催化的開發應用具有重要意義。
[1]Horvath I T, Rabai J. Facile catalyst separation without water:Fluorous biphase hydroformylation of olefins[J].Science,1994,266:72-75.
[2]Katarzyna S. Behaviour of the fluorocarbon surfactants in the monolayer at the water-air interface and in the bulk phase[J].Journal of Fluorine Chemistry,2013,150:109-116.
[3]Hongren C. Fluorocarbon and hydrocarbonN-Heterocyclic (C5-C7) imidazole-based liquid crystals[J]. Chem Asian J,2014,9(12):3418-3410.
[4]Xi Z, Dong M H. Fluorous Hydrogenation[J].Top Curr Chem,2012(308):233-246.
[5]Richter B, Spek A L, Van K G,etal. Fluorous versions of wilkinson’s catalyst:Activity in fluorous hydrogenation of 1-alkenes and recycling by fluorous biphasic separation[J].J Am Chem Soc,2000,122:3945-3951.
[6] 盧法冠. 氟代席夫堿體系中C—F鍵和C—H鍵的選擇性活化和功能化[D].濟南:山東大學,2014.
[7]伍新燕,吳成泰. 含氮雜冠醚結構單元的吲哚啉螺苯并吡喃的合成和性質[J].應用化學,1998,15(2):106-108.
[8]張丹,常曉紅,張兵,等. 金屬酞菁衍生物催化的模擬酶反應研究進展[J].遼寧大學學報自然科學版,2003,30(1):87-97.
[9]付時雨,詹懷宇,余惠生. 苯基羥胺及N-酰基衍生物的合成表征[J].華南理工大學學報,2000,28(5):59-63.
[10]張春春. 杯芳烴異羥肟酸的設計合成及過渡金屬配合物的仿酶功能研究[D].成都:四川大學,2003.
[11]呂志鳳,李鴻波. 異羥肟酸過渡金屬配合物的合成及其催化氧化性能研究[J].四川大學學報,2000,37(3):426-431.
[12]秦川,梁伍. 鹵代異羥肟酸鈷(II)配合物催化氧化二甲苯的研究[J].分子催化,2009,23(1):62-65.
[13]侯韓鵬,孫霞. 氧化鋁負載Co基F-T合成催化劑還原反應[J].分子催化,2011,25(3):238-243.
[14]杜治平,周彬. Cu(phen)Cl2催化甲醇氧化羰基化合成碳酸二甲酯[J].催化學報,2012,33(4):736-742.
[15]王小麗,徐南平,楊剛,等. 枝狀鈷配合物的合成及其對苯乙烯環氧化的催化性能[J].南京工業大學學報(自然科學版),2014,36(1):23-27.
Synthesis of Fluoro-phenyl Hydroxamic Acid Cobalt and Its Catalytic Activity for the Oxidation of Ethylbenzene
CHEN Xiao-feng,QIU Tao*,Lü Xin-yu
(Institute of Design and Research, Changzhou University, Changzhou 213164, China)
p-Perfluorohexyl nitrobenzene(1) andp-perfluorohexyl acid(3) were synthesized by reaction of perfluorobutyl iodide with 4-iodonitrobenzene and 4-iodobenzoic acid respectively. Fluoro-phenyl hydroxamic acid (5) was prepared by condensation reaction of the reduction product of 1 with acyl chloride product of 3. The hydroxamic aic-containing cobalt perfluorohexyl chain complexes(6) was synthesized by complexation reaction of 5. The structure was characterized by UV-Vis,1H NMR, FT-IR and HR-MS(EI). Its catalytic activity for the oxidation of ethylbenzeneIt was investgated. The results indicated that the amount of catalyst was 0.04 mmol and the reaction was carried out at 60 ℃ for 6 h, the conversion of ethyl benzene was up to 49.2% and the selection of acetophenone was 88.3% under the conditions of perfluorohexane as solvent. The selectivity of catalyst can maintain good after recycling for five times.
fluoro-phenyl hydroxamic acid cobalt; fluorocarbon catalysis; oxidation of ethyl benzene; synthesis; catalytic activity
2015-11-12;
2016-06-16
國家自然科學基金資助項目(21276030)
陳小峰(1990-),男,漢族,江蘇淮安人,碩士研究生,主要從事精細化工的研究。E-mail: xfchen2009@163.com
邱滔,研究員, Tel. 0519-86330181, E-mail: qiutao@cczu.edu.cn
O641.4; O621.3
ADOI: 10.15952/j.cnki.cjsc.1005-1511.2016.09.15376
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
