極性反轉一鍋法制備聯芳基七元內酯
2016-09-13 07:19:16解卉旖徐莉梅趙錦妮杜文婷
浙江化工 2016年8期
關鍵詞:催化劑
解卉旖,徐莉梅,趙錦妮,杜文婷
(杭州醫學院,浙江 杭州 310053)
極性反轉一鍋法制備聯芳基七元內酯
解卉旖,徐莉梅,趙錦妮,杜文婷*
(杭州醫學院,浙江杭州310053)
以N-雜環卡賓做極性反轉催化劑,以無水碳酸鉀為堿、18-冠-6為相轉移催化劑,給氧條件下一鍋法合成了一系列聯芳基七元內酯。該方法適用于各種結構類型的底物;相對于傳統的聯芳基七元內酯合成方法來說,具有操作簡單、一步反應、條件溫和,對環境相對友好及較高產率等優點,為聯芳基七元內酯的合成提供了一種實用并具有原子經濟性的途徑。
聯芳基七元內酯;N-雜環卡賓;極性反轉;一鍋法
聯芳基七元內酯是一種具有抗心律失常活性的生物活性化合物,它也是許多多功能環氧樹脂的重要結構部分,是各種粘合劑、密封膠、覆膠的組成成分,是某些化工產品的必要原料[1],因此探索合成聯芳基七元內酯的方法引起有機與藥物化學工作者的關注。近年來,已有多篇文獻報道聯芳基七元內酯的多種合成方法[2],包括聯芳基二醇或者二醛的氧化內酯化、聯芳基烯烴的氧化斷裂、聯芳基二羧酸的還原等等,見Scheme 1。這些合成方法存在以下技術缺陷:需用貴重稀缺或難以制備的金屬催化劑、毒性/強腐蝕性試劑,反應過程污染嚴重,反應條件苛刻(無水無氧、加壓、高溫、長時間反應等)和后處理繁瑣及產率低等。即使是Gerhard B報道的分子內偶聯方法[2h],也需要以鎳復合物和三苯基磷催化并需要丁基鋰試劑參與。
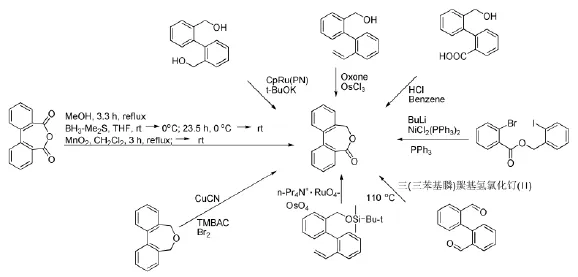
Scheme 1
極性反轉的有機催化反應打破了傳統意義上對于合成子反應性能的理解,開辟了一條用非傳統方法構建碳碳鍵的途徑,成為有機化學家用來改變常規反應模式的常用方法。隨著對極性反轉機理理解的加深,極性反轉催化劑被不斷開發、應用,氰化物由于其劇毒特性漸漸淡出在極性反轉有機催化中的應用,N-雜環卡賓(NHCs)由于其性質獨特,種類繁多且毒性較小等優點而成為該類反應中最常用的有機小分子催化劑之一。用于極性反轉的NHCs最常見的應用有Benzoin反應、Stetter反應、酯交換反應、開環反應等。本文報道的N-雜環卡賓(NHCs)通過極性反轉參與促進的聯芳基七元內酯的合成新方法,與NHCs介導的酯交換反應有本質差別,其生成機理也會截然不同。另一方面,N-雜環卡賓的二聚體往往因為化學不穩定性而難以分離得到,我們在研究過程中成功分離得到兩種N-雜環卡賓二聚體。
1 實驗部分
1.1儀器與試劑
核磁共振采用Brüker AM型(中國,杭州)核磁共振儀測定,三甲基硅烷為內標。元素分析采用Carlo ERBA-1108型元素分析儀器。質譜采用Finnigan LCQ DECA光譜分析儀測定。薄層層析(TLC)和柱層析分別使用山東青島海洋化工廠生產的薄層層析硅膠GF254型硅膠和300~400目柱層析硅膠。實驗中所用無水乙腈和18-冠-6由百靈威科技公司購置。原料1和催化劑I,II,III自制獲得。
1.2實驗方法
1.2.1聯芳基七元內酯2的合成
向干燥的反應管中依次加入化合物1(0.2mmol)、N-雜環卡賓噻唑鹽III(0.2 mmol)和無水乙腈4 mL,攪拌下加入無水碳酸鉀(0.4 mmol)和18-crown-6(0.12 mmol)。用氧氣置換反應體系中的空氣,氧氣下(氣球)室溫反應約3 h,待原料反應完全后停止反應,以旋轉蒸發儀蒸干溶劑,柱層析(乙酸乙酯:石油醚 =1:40)分離,得產物2-聯苯[c,e]氧雜卓-5(7H)-酮2a:m.p.131℃~134℃;1H NMR(400 MHz,CDCl3)δ 8.02(dd,J=7.8,1.2 Hz,1H),7.73~7.62(m,3H),7.60~7.52(m, 2H),7.51~7.43(m,2H),5.05(d,J=24.0 Hz,2H)。13C NMR(100 MHz,CDCl3)δ 169.4,138.8,135.5,134.4,132.9,131.4,129.8,129.1,128.8,128.7,127.5,126.9,122.3,72.8.LRMS:[M]+,210.1;Anal.Calcd for C14H10O2(210):C,80.34;H,5.39;O,14.27.Found:C,80.26;H,5.41;O,14.20。
3-溴代聯苯 [c,e]氧雜卓-5(7H)-酮2b:1H NMR(400 MHz,DMSO)δ 8.00(d,J=8.0 Hz,1H),7.85~7.77(m,3H),7.50~7.46(m,3H),5.50(d,J=9.0 Hz,1H),5.48(d,J=9.0 Hz,1H).LRMS:[M]+,287.9。
9-溴代聯苯 [c,e]氧雜卓-5(7H)-酮2c:1H NMR(400 MHz,DMSO)δ 8.43(d,J=8.0 Hz,1H),7.68~7.54(m,6H),5.51(d,J=9.0 Hz,1H),5.44(d,J=9.0 Hz,1H).LRMS:[M]+,288.0。
3,9-二溴代聯苯[c,e]氧雜卓-5(7H)-酮2d:1H NMR(400 MHz,DMSO)δ 8.38(d,J=7.0 Hz,1H),7.84~7.59(m,6H),5.50(d,J=9.0 Hz,1H),5.43(d,J=9.0 Hz,1H).LRMS:[M]+,367.9。
3,9-二硝基聯苯[c,e]氧雜卓-5(7H)-酮2e:1H NMR(400 MHz,DMSO)δ 8.40(s,1H),8.31~8.24(m,3H),8.05(d,J=7.4 Hz,1H),7.61 (d,J=7.4 Hz,1H),5.36(d,J=9.0 Hz,1H),5.26(d,J=9.0 Hz,1H).LRMS:[M]+,300.0。
3,9-二甲氧基聯苯[c,e]氧雜卓-5(7H)-酮2f:1H NMR(400 MHz,DMSO)δ 7.88~7.70(m,3H),7.23(dd,J=7.5,1.5 Hz,1H),7.12(d,J =1.5 Hz,1H),7.02(dd,J=7.5,1.5 Hz,1H),5.17(d,J=9.0 Hz,1H),5.07(d,J=9.0 Hz,1H),4.09(s,3H),4.01(s,3H).LRMS:[M]+,270.1。
3,9-二甲基聯苯[c,e]氧雜卓-5(7H)-酮2g:1H NMR(400 MHz,DMSO)δ 7.68-7.62(m,3H),7.41~7.38(m,3H),5.05(d,J=9.0 Hz,1H),4.92(d,J=9.0 Hz,1H),2.40(s,3H),2.396(s,3H).LRMS:[M]+,238.1。
3,10-二甲氧基聯苯[c,e]氧雜卓-5(7H)-酮2h:1H NMR(400 MHz,DMSO)δ 7.85~7.68(m,3H),7.23(dd,J=7.5,1.5 Hz,1H),7.13(d,J=1.5 Hz,1H),7.01(dd,J=7.5,1.5 Hz,1H),5.52(d,J=9.0 Hz,1H),5.43(d,J=9.0 Hz,1H),3.83(s,3H),3.77(s,3H).LRMS:[M]+,270.1。
3,10-二甲基聯苯[c,e]氧雜卓-5(7H)-酮2i:1H NMR(400 MHz,DMSO)δ 7.67~7.62(m,3H),7.45~7.37(m,3H),5.06(d,J=9.0 Hz,1H),4.93(d,J=9.0 Hz,1H),2.46(s,3H),2.43(s,3H).LRMS:[M]+,238.1。
二萘[2,1-c:1',2'-e]氧雜卓-3(5H)-酮2j:1H NMR(400 MHz,DMSO)δ 8.24(d,J=8.0 Hz,1H),7.96(d,J=8.0 Hz,1H),7.65~7.59(m,3H),7.37~7.32(m,5H),7.12-7.10(m,1H),6.99~6.98(m,1H),5.24(d,J=9.0 Hz,1H),5.12(d,J=9.0 Hz,1H).LRMS:[M]+,310.1。
1.2.2N-雜環卡賓二聚體3和4的合成
向干燥的反應管中依次加入2’-(溴甲基)聯苯基-2-甲醛(1a)55 mg(0.2 mmol)、N-雜環卡賓噻唑鹽I 31.4 mg(0.1 mmol)和無水乙腈4 mL,攪拌下加入氫化鈉16.0 mg(0.4 mmol)。用氧氣置換反應體系中的空氣,氧氣下(氣球)室溫反應約3 h,待原料反應完全后停止反應,以旋轉蒸發儀蒸干溶劑,柱層析(乙酸乙酯:石油醚 =1:40)分離,得產物2a 13.5 mg收率為33%;同時柱層析(二氯甲烷:甲醇 =30:1)得到N-雜環卡賓二聚體(3)8.4 mg,收率為9%:1H NMR(400 MHz,CDCl3)δ 7.38~7.30(m,6H),7.24~7.21(m,4H),4.95(s,4H),3.77(t,J=6.6 Hz,4H),2.74(t,J=6.2 Hz,4H),2.01(s,6H).LRMS:[M]+,466.2。
向干燥的反應管中依次加入2’-(溴甲基)聯苯基-2-甲醛(1a)55 mg(0.2 mmol)、N-雜環卡賓噻唑鹽 III 25.5 mg(0.1 mmol)和無水乙腈4 mL,攪拌下加入叔丁醇鉀44.9 mg(0.4 mmol)。用氧氣置換反應體系中的空氣,氧氣下(氣球)室溫反應約3 h,待原料反應完全后停止反應,以旋轉蒸發儀蒸干溶劑,干法上樣柱層析(乙酸乙酯:石油醚 =1:40)分離,得產物2a 24.4 mg,收率為58%,同時柱層析(乙酸乙酯:石油醚 =1:4)得到N-雜環卡賓二聚體 (4)6.9 mg,收率14%:1H NMR(400 MHz,CDCl3)δ 3.25(s,6H),2.10(s,6H),2.06(s,6H).LRMS:[M]+,254.1。
2 結果與討論

Table 1對聯芳基七元內酯合成的反應條件篩選優化a
借鑒前期對NHCs極性反轉催化下的α-芳基酮和芳香羧酸芐酯的合成經驗[3],我們有針對性地選擇了卡賓鹽、堿和溶劑進行了篩選優化(Table 1)。以卡賓鹽I為催化劑,乙腈為溶劑,對具有代表性的堿K2CO3、NaH和t-BuOK進行篩選優化,發現碳酸鉀更有利于目標產物的生成(Entries 1,2,6);而碳酸鉀與相轉移催化劑18-冠-6配合,可使產率得到顯著提高(Entries 9,10)。對溶劑的篩選優化結果,類似于NHC參與的醛與鹵代烴的親核取代反應[3],仍舊是無水乙腈略勝一籌(Entries 3-5);卡賓鹽I、II和III(50 mol%)在相同堿和溶劑條件下分別參與反應,結果顯示III得到的聯芳基七元內酯明顯高于I和II(Entries 6-8)。在Entry 2與Entry 3記錄的反應中,意外的分別分離得到了催化劑N-雜環卡賓的二聚體3與4(Scheme 2),證明了醛基與溴甲基反應過程中,卡賓鹽到NHCs的轉化率過低,部分卡賓鹽自身反應生成了二聚物,從而無法實現催化循環;這也是當卡賓鹽I由50 mol%投料量變成100 mol%投料量,產率發生顯著提高的重要原因(Entry 3,9)。從而我們初步確定了最佳反應體系:以卡賓鹽III化學計量參與反應,碳酸鉀配合相轉移催化劑18-冠-6,在無水乙腈溶劑中室溫、給氧,可高效生成聯苯七元內酯。

Scheme
在上述最佳反應體系下,我們應用不同取代的鹵芐聯苯基甲醛和溴甲基聯萘甲醛合成聯芳基七元內酯衍生物,結果表明,在優化條件下可以得到65%~92%的產率(Table 2)。該方法適用于聯芳基七元內酯的合成。

Table 2 NHCs促進聯芳基七元內酯的合成方法的底物拓展

aThe reaction was performed under oxygen,with 1 (0.2 mmol)and 100 mol%of III,in the presence of anhydrous K2CO3(2 equiv)and of 18-crown-6(0.6 equiv)in 4 mL of anhydrous CH3CN.
3 結論
給氧條件下,以無水乙腈為溶劑,以卡賓鹽III為催化劑,以無水碳酸鉀為堿、18-冠-6為相轉移催化劑,對卡賓鹽進行現場游離,室溫攪拌反應約3 h,一鍋法合成了一系列聯芳基七元內酯衍生物。本方法屬分子內偶聯,但產物的生成機理依靠NHCs的極性反轉,與傳統的分子內偶聯截然不同,相對于傳統的聯芳基七元內酯合成方法來說,具有操作簡單、一步反應、條件溫和,且避免使用金屬催化劑及苛刻條件,對環境相對友好等優點,以100 mol%噻唑類卡賓鹽參與反應可獲得高產率,為聯芳基七元內酯的合成提供了一條實用并具有原子經濟性的途徑。
[1](a)Samvelyan V M,Vasilyan S S.Antiarrhythmic properties of some cholinolytic substances[J].Krovoobrashchenie,1981,14:13-18.(b)Atsushi S,Takeshi E,Akane S. Low shrinkage epoxy-cationic curable compositions:WO,2007135094 A1[P].2007-11-29.(c)Ralf R,Richard P K.N-Heterocycles with a 2-azaallyl system.5.7-Methoxyand 7-alkylthio-5H-dibenz[c,e]azepinium salts:7-membered N-heterocycles with an integrated 2-azaallyl system [J].Chemiker-Zeitung,1991,115:193-201.
[2](a)Wu X F.A general and efficient zinc-catalyzed oxidation of benzyl alcohols to aldehydes and esters[J].Chemistry-A European Journal,2012,18:8912-8915.(b)Makoto B E,Yuto S,Haruhiko F,et al.Highly efficient synthesis of medium-sized lactones via oxidative lactonization:concise total synthesis of isolaurepan[J].Org.Biomol. Chem.,2010,8:39-42.(c)Sohei O,Takahide F,Yuji M,et al.Hydroruthenation triggered catalytic conversion of dialdehydes and keto aldehydes to lactones[J].Chem. Commun.,2009,(44):6741-6743.(d)Ito M,Osaku A,Shiibashi A,et al.An efficient oxidative lactonization of 1,4-diols catalyzed by CpRu(PN)complexes[J].Org. Lett.,2007,9:1821-1824.(e)Edwards D J,House D,Sheldrake H M et al.Kinetic and thermodynamic control of axial chirality in biaryl-derived fused oxazolidine lactams exploiting a centre-axis relay of unit efficiency[J].Organic&Biomolecular Chemistry,2007,5:2658-2669.(f)Whitehead D C,Travis B R,Borhan B.The OsO4-mediated oxidative cleavage of olefins catalyzed by alternative osmium sources[J].Tetrahedron Lett.,2006,47:3797-3800.(g)Schomaker J M,Travis B R,Borhan B.Direct lactonization of alkenols via osmium tetroxide-mediated oxidative cleavage[J].Org.Lett.,2003,5:3089-3092.(h)Gerhard B,Jürgen H,Petra H,et al.Novel concepts in directed biaryl synthesis.Part 90.Synthesis of constitutionally unsymmetric 7-membered biaryl lactones by Nimediated intramolecular coupling[J].Synlett 2000,(12):1822-1824.
[3] (a)Li Y,Du W,Deng W P.NHCs-mediated benzoates formation directly from aromatic aldehydes and alkyl halides[J].Tetrahedron,2012,68:3611-3615.(b)Lin L,Li Y,Du W,et al.The NHCs-mediated cross-coupling of aromatic aldehydes with benzyl halides:synthesis of α-aryl ketones[J].Tetrahedron Lett.,2010,51:3571-3574.
A One-pot Synthesis of Biaryl Seven-membered Lactones via Umpolung Reaction
XIE Hui-yi,XU Li-mei,ZHAO Jin-ni,DU Wen-ting*
(Hangzhou Medical College,Hangzhou,Zhejiang 310053,China)
To develop a simple and efficient procedure for the synthesis of biaryl seven-membered lactones.The umpolung reaction was promoted by N-heterocyclic carbenes under oxygen,with anhydrous potassium carbonate as base and with 18-crown-6 as a phase-transfer catalyst.The practical protocol was found to be compatible with different structurally diverse substrates.This synthesis method has the advantage of operation simplicity,one-step reaction,mild condition,relative environment-friendly and moderate to high yields,providing a useful and atom-economic approach to the synthesis of biaryl seven-membered lactones.
biaryl seven-membered lactones;N-heterocyclic carbine;umpolung reaction;one-pot
1006-4184(2016)8-0030-05
2016-03-18
2016年度浙江省自然科學基金一般項目 (LY16H300004),2016年浙江省大學生科技創新活動計劃 (新苗人才計劃,2016R436005),2014年度浙江省衛生高層次創新人才項目。
解卉旖(1996-),女,浙江金華人,在讀專科生。
杜文婷,女,博士,副教授。E-mail:ddwwtt@163.com。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
