石墨爐法檢測螺旋藻片中鉛的干擾研究及消除
2016-09-02 08:28:15張華珺孫亮楊玲鄔國慶北京市藥品檢驗所北京102206
食品研究與開發 2016年13期
關鍵詞:標準
張華珺,孫亮,楊玲,鄔國慶(北京市藥品檢驗所,北京102206)
?
石墨爐法檢測螺旋藻片中鉛的干擾研究及消除
張華珺,孫亮,楊玲,鄔國慶*
(北京市藥品檢驗所,北京102206)
研究分析干擾螺旋藻片中鉛含量測定的影響因素,依據樣品特點及基體改進劑作用機理,選擇硝酸鈀、硝酸鎂、抗壞血酸作為基體改進劑協同使用,優化試驗條件為:2 g/L硝酸鈀溶液、1 g/L硝酸鎂溶液與5%抗壞血酸溶液以3∶3∶4(體積比)濕法添加,灰化溫度提高至1200℃,原子化溫度1900℃,實現了螺旋藻片中鉛含量的準確定量。
螺旋藻;石墨爐法;鉛檢測;干擾消除
螺旋藻分布在熱帶、亞熱帶地區。作為地球上最古老的物種之一,被人類食用已有100多年的歷史。螺旋藻中的極大螺旋藻、鈍頂螺旋藻兩個品種允許作為保健食品的原料。螺旋藻中蛋白質含量比例較高,還含有豐富的類胡籮卜素和B族維生素及礦物質,具有較高的營養價值,近年來被廣泛用于臨床研究[1-2]。但有研究表明[3-4],螺旋藻因富含多糖、蛋白質和脂類等大分子物質可以提供大量的基團吸附重金屬,因而對鉛具有一定程度的富集作用。而鉛是人類生存環境中普遍存在的重金屬,從1997年衛生部批準的第一個螺旋藻片劑保健食品開始,我國以藻類為原料的固體飲料和膠囊產品鉛指標限量始終按照2.0 mg/kg的標準執行。按照GB 16740-2014《食品安全國家標準保健食品》中規定[5],在日常監管工作中,應采用GB 5009.12-2010《食品安全國家標準食品中鉛的測定》[6]石墨爐原子吸收光譜法使用磷酸二氫銨作為基體改進劑(NH4H2PO3法)測定螺旋藻保健食品中有毒重金屬鉛的含量,但在實際工作中我們觀察試驗現象,發現測定結束后石墨管的鉛記憶效應增強不易消除,有殘渣附著在石墨管內壁上。考慮到螺旋藻具有較大的比表面積因而對鉛應具有較大的吸附親和力,但鉛的測定結果又比較低,故我們分析推斷可能存在基質干擾降低了鉛的吸收信號。
試驗通過分析螺旋藻片的組成成分,研究了鉛測定的干擾因素,依據基體改進劑的作用機理[7],篩選硝酸鈀、硝酸鎂和抗壞血酸作為混合基體改進劑聯合使用,使鉛形成熱穩定的合金不在灰化階段損失同時利用抗壞血酸在石墨爐中形成富碳環境,加速一些金屬氧化物分解生成原子,繼而通過提高灰化溫度,去除基質成分對鉛的測定干擾,實現了螺旋藻片中重金屬鉛的準確定量。
1 試驗部分
1.1材料與儀器
硝酸(BV-Ⅲ級):北京化工廠;30%過氧化氫(MOS級):北京化學工業研究所;硝酸鈀:Alfa Aesar公司;硝酸鎂(分析純):北京雙環化學試劑廠;抗壞血酸:Sigma公司。
0.5mol/L硝酸溶液、2 g/L硝酸鈀溶液(含4%硝酸)、1 g/L硝酸鎂溶液與5%抗壞血酸溶液。
標準對照品:GBW(E)080129鉛單元素溶液標準溶液(100 mg/L),中國計量科學研究院。
螺旋藻標準物質:[GBW10025-GSB-16螺旋藻生物成分標準物質,含鉛(2.8±0.2)μg/g]:地球物理地球化學勘查研究所。
樣品:螺旋藻片(2種,不同品牌,樣品編號1、2)。
AND GR-202電子天平,精密度0.01 mg:A&D Company;微波消解儀:德國MILESTONE ETHOS 1;原子吸收光譜儀,帶GF95Z型石墨爐,FS95型自動進樣器:美國Thermo AA-M6-MK2;鉛空心陰極燈:美國Thermo。
1.2方法
1.2.1樣品制備
將樣品研細后精密稱取0.5 g(精確至0.000 1 g),置于聚四氟乙烯消解杯中,同時做試劑空白,加入8 mL硝酸,輕輕搖晃使樣品粉末分散于硝酸中,加蓋,靜置過夜后,加入30%過氧化氫2 mL,于100℃電熱板上加熱20 min,冷卻后,上消化罐,進行微波消解。消解完畢將盛有消解液的消解杯置于100℃電熱板上加熱趕酸至溶液近干,冷卻后用少量超純水多次潤洗消解杯,合并洗滌液并轉移至10 mL容量瓶中,用超純水定容至刻度,混勻,備用。
1.2.2標準曲線的建立
精密吸取100 mg/L的鉛元素標準溶液1 mL,加0.5 mol/L硝酸溶液稀釋定容至100 mL(每1 L含鉛1 mg)。精密量取1 mg/L的標準鉛溶液1 mL,至25 mL容量瓶中,加0.5 mol/L硝酸溶液稀釋至刻度,混勻,得40 μg/L鉛標準主溶液,由自動進樣器分別吸取40 μg/L鉛標準使用液0.0、1.0、3.0、5.0、10.0、15.0 μL,用0.5mol/L硝酸溶液稀釋至20 μL,得0.0、2.0、6.0、10.0、20.0、30.0 μg/L的標準曲線。
石墨爐升溫程序見表1。
按照表1所示優化好的石墨爐升溫程序,濕法添加2 g/L硝酸鈀溶液3 μL、1 g/L硝酸鎂溶液3 μL、5%(w/v)抗壞血酸溶液4 μL作為基體改進劑,進樣總體積為30 μL,Zeeman扣背景,吸收信號以峰高表示。以吸收信號為縱坐標,濃度C(μg/L)為橫坐標進行一次線性擬合標準曲線。結果表明,鉛濃度在(0~30)μg/L范圍內線性關系良好,其回歸方程為:A=7.87×10-3C+ 1.01×10-2,相關系數:0.999 0,重復測定空白溶液11次,用3σ除以標準曲線斜率計算得檢出限為0.1 μg/L。
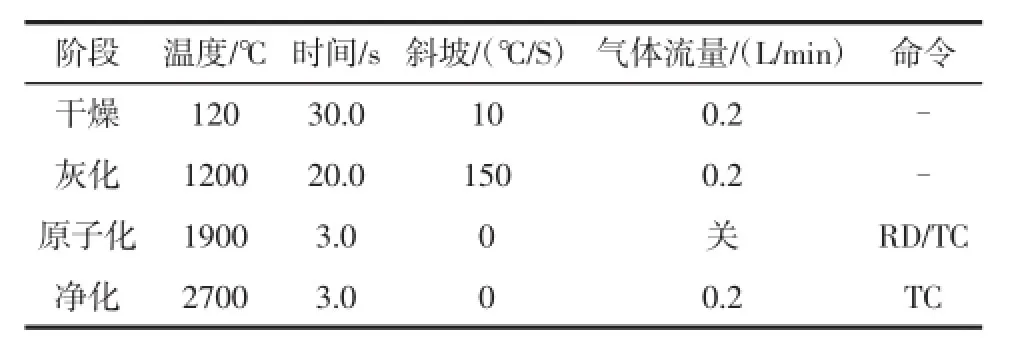
表1 石墨爐升溫程序Table 1 Furnace heating program
2 結果分析
2.1干擾因素確定
考慮到石墨爐法高溫原子化采用直接進樣程序升溫方式,如果灰化階段不能有效凈化基體,原子化階段的基體成分則會如自由原子一般,不僅濃度高、而且停留時間也長,極易造成基體干擾和背景吸收。故試驗中選擇加標回收試驗判斷是否存在基體干擾,選擇鉛分析波長283.3 nm的鄰近的非吸收波長280.2 nm作為甄別背景吸收是否存在的判定波長,若有信號則表明測鉛時有背景吸收。
試驗中分別使用NH4H2PO3法和新建方法對20 μg/L鉛標準溶液、樣品1、樣品2進行回收率分析,測定結果見表2。
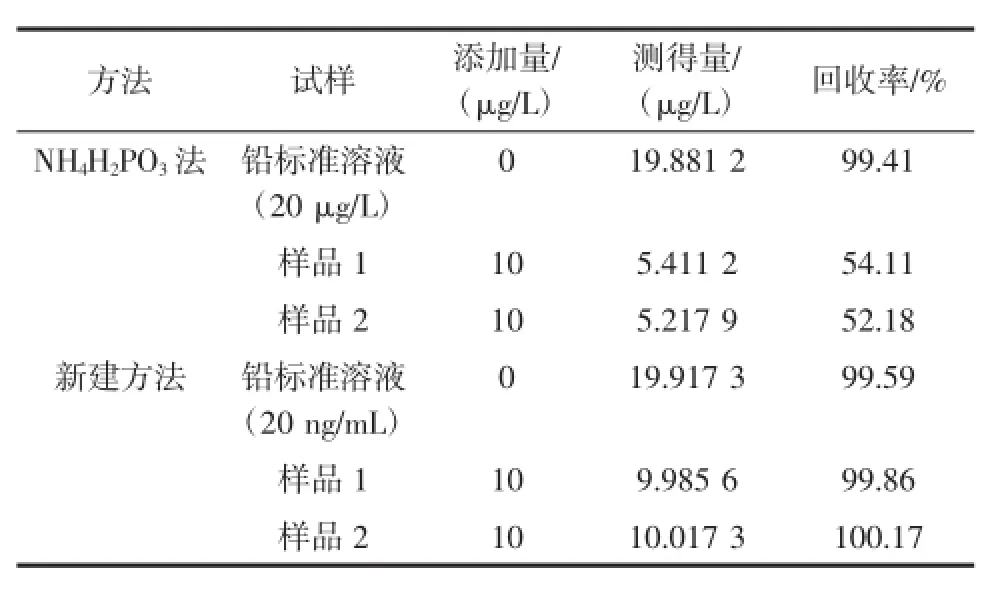
表2 基體干擾的判定Table 2 The estimate of substrate interference
使用NH4H2PO3法和新建方法測得鉛標準溶液的回收率一致,結果良好,但兩種方法用于測定樣品時,新建方法消除基體干擾效果良好,NH4H2PO3法測得樣品回收率約為新建方法的50%,說明NH4H2PO3法確實存在基體干擾造成了分析元素鉛的信號降低,推測原因為,螺旋藻中富含多種營養元素,組成成分復雜,待分析元素鉛有可能與基體中的共存物形成化合物,從而對鉛測定產生基體干擾。
試驗中考察了20 μg/L鉛標準溶液、樣品1、樣品2分別按照NH4H2PO3法和新建方法,使用鉛的非吸收波長280.2 nm測定是否有背景吸收,測定結果見表3。
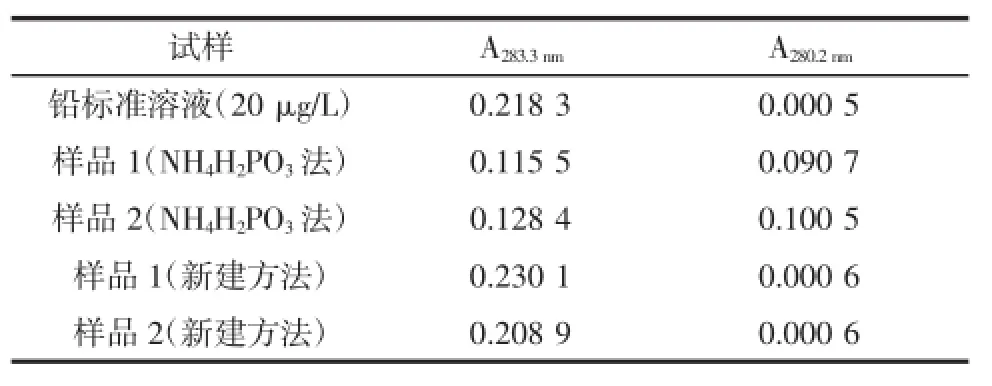
表3 背景吸收Table 3 Background absorption
如表3所示,NH4H2PO3法存在背景吸收干擾,分析其干擾來源,認為基體有可能在原子化階段以“氣態分子、鹽微粒、煙霧”等形式存在于石墨管中從而產生分子吸收、光散射等背景吸收。
2.2基體改進劑的篩選
石墨爐法分析鉛時,基體干擾主要發生在于灰化和原子化階段,使用基體改進劑的目的在于使基體形成易揮發的化合物在灰化階段去除;或降低待測元素的揮發性以防止在灰化過程中損失。
由于基體改進劑協同使用在扣除背景干擾,減少分析誤差,提高方法準確性和適用性方面較單一基體改進劑更為理想,試驗中通過考察待測樣品原子化峰形,加標回收率及螺旋藻成分分析標準物質的測定值與標準值的差異,對比分析了3種混合基體改進劑去除基體干擾的效果,結果發現:3種基本改進劑作用下的樣品原子化峰形均尖銳光滑對稱,但只有硝酸鈀、硝酸鎂和抗壞血酸作為混合基體改進劑聯合使用測定螺旋藻片中鉛的加標回收率結果良好,螺旋藻成分分析標準物質的測定值在標準值規定范圍內。
故試驗中最終確定硝酸鈀、硝酸鎂和抗壞血酸作為混合基體改進劑聯合使用,使鉛形成熱穩定的合金不在灰化階段損失同時利用抗壞血酸在石墨爐中形成富碳環境,加速一些金屬氧化物分解生成原子,從而可通過提高灰化溫度,去除樣品基質的干擾,達到避免灰化損失和改善背景吸收干擾的目的。
2.3灰化,原子化溫度優化
灰化溫度在1 200℃時,吸光度有最大吸收值;高于1 200℃吸光度急劇下降,故選擇的灰化溫度為1 200℃,在此溫度下樣品中大部分干擾成分被凈化;原子化溫度從1900℃始樣品中的鉛的吸光度增加趨勢不明顯,說明1900℃即可使樣品中的鉛充分原子化。
2.4精確度試驗
兩種樣品各取5份,按1.2進行平行性試驗測定,測得結果見表4。

表4 精密度試驗結果Table 4 The test results of accuracy(n=5)
相對標準偏差<5%,滿足GB 5009.12-2010《食品安全國家標準食品中鉛的測定》中規定石墨爐法精密度應≤20%的方法要求。
2.5回收率試驗
為了進一步檢驗方法精確度,采用添加鉛元素標準溶液于樣品中進行加標回收率測定,每種樣品做3份。試驗結果如表5所示,采用此法測得的回收率在94%~102%之間,符合質控要求。

表5 回收率試驗結果Table 5 The test results of the recovery experiment(n=3)
2.6標準物質質控試驗
為進一步驗證分析方法的準確性和基體改進劑消除基質干擾的效果,試驗中,精密稱取0.1 g(精確至0.0001 g)GBW10025-GSB-16螺旋藻生物成分標準物質,按1.2制備,用水定容至25 mL容量瓶中,搖勻得質控樣品溶液,平行制備5份。標準物質試驗結果見表6。

表6 標準物質試驗結果Table 6 The test results of the reference material(n=5)
由于石墨爐原子吸收光譜法屬于相對測量,在測定過程中同時引入螺旋藻標準物質作為方法驗證使用,通過5次測定值之間的離散程度,及其測定值與標準值的差異可以直觀的反映儀器工作狀態和測定方法的精確度[8]。
2.7樣品測定及不同方法對比
為考察方法的適用范圍及準確度,試驗中分別選擇硬膠囊、片劑、粉劑3種以螺旋藻作為主要原料的常見保健食品劑型,同時運用本法、NH4H2PO3法和電感耦合等離子體原子發射光譜法(ICP法)[9]進行鉛含量測定,見表7。

表7 樣品含量測定結果Table 7 Analytical results of samples(n=3)
3 結論
本文依據基體改進劑的作用機理,通過分析確認干擾螺旋藻片中鉛含量測定的影響因素,優化建立了一種新型基體改進劑組合,可用于石墨爐原子吸收光譜法檢測以螺旋藻為主要原料的保健食品中鉛的含量,并采用ICP法對NH4H2PO3法和文章中新建方法的測定結果進行驗證,結果表明:新建方法鉛含量測定結果準確,可有效去除螺旋藻中其他成分對鉛的測定干擾,國內外均未見相似報道,新建方法具有創新性。
[1]王文博,高俊蓮,孫建光,等.螺旋藻的營養保健價值及其在預防醫學中的應用[J].中國食物與營養,2009(1):48-51
[2]張文,吳清平,吳軍林.螺旋藻營養保健價值及開發應用進展[J].食品與發酵科技,2013,49(3):89-92
[3]龔仁敏,陳發揚,劉必融,等.天然和預處理的極大螺旋藻粉對鉛的吸附[J].南京農業大學學報,2004,27(3):107-110
[4]Roy D,Greenlaw P N,Shane B S.Adsorption of heavy metals by green algae and ground rice hulls[J].JEnvironSciHealthA,1993(28): 37-50
[5]中華人民共和國國家衛生和計劃生育委員會.GB 16740-2014食品安全國家標準保健食品[S].北京:中國標準出版社,2014:1-2
[6]中華人民共和國衛生部.GB 5009.12-2010食品安全國家標準食品中鉛的測定[S].北京:中國標準出版社,2010:1-10
[7]鄧勃,何華焜.原子吸收光譜分析[M].北京:化學工業出版社,2003: 280-284
[8]盧曉華.標準物質在化學測量結果不確定度評定中的應用舉例[J].中國計量,2008(1):68-69
[9]國家藥典委員會.中華人民共和國藥典四部[M].北京:中國醫藥科技出版社,2015:42-43
Research for the Elimination of Matrix Interference on the Determination of Lead in Spirulina Tablets by GFAAS
ZHANG Hua-jun,SUN Liang,YANG Ling,WU Guo-qing*
(Beijing Institute For Drug Control,Beijing 102206,China)
The interfering factors in determination of lead in Spirulina tablets were analyzed,based on sample characteristics and the matrix modifier chemical mechanism,we selected palladium nitrate,magnesium nitrate and ascorbic acid as matrix modifier.The optimize conditions were as follows:added palladium nitrate solution 2 g/L,magnesium nitrate solution 1 g/L and ascorbic acid solution 5%:a proportion of 3∶3∶4(volume ratio),ashing temperature to 1 200℃,atomization temperature 1 900℃,achieved the accurately quantitative in spirulina tablets lead content determination.
spirulina tablets;GFAAS;lead testing;the elimination of interference
10.3969/j.issn.1005-6521.2016.13.034
張華珺(1981—),女(漢),主管藥師,碩士研究生,研究方向:保健食品與化妝品檢測與安全評價。
2016-03-05
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
