無(wú)機(jī)氯化物熔鹽在乏燃料干法后處理中的應(yīng)用進(jìn)展*
2016-08-31 08:43:22王有群何輝林如山葉國(guó)安唐洪彬賈艷虹
無(wú)機(jī)鹽工業(yè) 2016年8期
關(guān)鍵詞:后處理
王有群,何輝,林如山,葉國(guó)安,唐洪彬,賈艷虹
(中國(guó)原子能科學(xué)研究院放射化學(xué)研究所,北京102413)
?
綜述與專論
無(wú)機(jī)氯化物熔鹽在乏燃料干法后處理中的應(yīng)用進(jìn)展*
王有群,何輝,林如山,葉國(guó)安,唐洪彬,賈艷虹
(中國(guó)原子能科學(xué)研究院放射化學(xué)研究所,北京102413)
綜述了無(wú)機(jī)氯化物熔鹽在乏燃料后處理中的應(yīng)用進(jìn)展,主要介紹了LiCl-KCl熔鹽在金屬乏燃料中的電解精煉和熔劑萃取、LiCl-Li2O在金屬氧化物或混合金屬氧化物(MOX)乏燃料中的電化學(xué)還原及NaCl-2CsCl在氧化物乏燃料電化學(xué)沉積中的應(yīng)用進(jìn)展。展望了中國(guó)無(wú)機(jī)氯化物熔鹽在干法后處理中的應(yīng)用前景及發(fā)展方向。
氯化物熔鹽;乏燃料;干法后處理
隨著快堆技術(shù)的發(fā)展,快堆乏燃料的處理在世界范圍內(nèi)得到越來(lái)越多的關(guān)注。FR乏燃料具有高燃耗、高輻照和高钚量等缺點(diǎn),使基于溶劑萃取的水法后處理流程難以滿足分離需要,而較適合采用干法后處理(Dry Reprocessing)技術(shù)。干法后處理技術(shù)主要包括氟化物揮發(fā)法(Fluoride Volatility Method,F(xiàn)VM)[1]、熔鹽電解法(Molten Salt Electrolysis)和熔鹽萃取法(Molten Salt Extraction)等。熔鹽電解法主要包括氧化物電化學(xué)沉積(Electrowinning)和電解精煉(Electro Refining,ER)2種方法。雖然氟化物揮發(fā)法具有去污系數(shù)高和產(chǎn)物純等特點(diǎn),但存在HF、F2、ClF3和ClF等氟化試劑性質(zhì)活潑、氟化反應(yīng)放熱劇烈及對(duì)設(shè)備要求高等不足,目前有俄羅斯[2]和捷克等國(guó)家開(kāi)展此方面的研究。
干法后處理中,氯化物熔鹽主要應(yīng)用于電化學(xué)還原和熔鹽電解法及熔鹽萃取等方面。目前,高溫熔鹽電解干法后處理研究的主要國(guó)家有美國(guó)、俄羅斯、歐盟、日本和韓國(guó)等。采用高溫氯化物熔鹽可處理包括金屬、氧化物及MOX氮化物和碳化物等乏燃料及Purex流程產(chǎn)生的高放廢液(HLLW)等。
熔鹽干法后處理的優(yōu)點(diǎn):1)采用無(wú)機(jī)氯化物熔鹽,耐輻射和耐熱性能優(yōu)異,可處理短冷卻時(shí)間、高燃耗和高钚的乏燃料[3],減少了復(fù)雜的核燃料運(yùn)輸過(guò)程;2)核燃料于熔鹽中的溶解性較好,適合處理各種乏燃料[2];2)與快堆或加速器驅(qū)動(dòng)次臨界核能系統(tǒng)(Accelerator Driven Subcritical System,ADS)結(jié)合,可減少長(zhǎng)壽命放射性廢物量[4];3)處理過(guò)程中無(wú)水,無(wú)中子慢化現(xiàn)象,可降低臨界危險(xiǎn)[3];4)最終廢物量小[3];6)防核擴(kuò)散性。與水法相比,干法后處理得到的元素的純度低。
通常采用LiCl-KCl、NaCl-KCl和NaCl-2CsCl等混合氯化物熔鹽作為介質(zhì)的原因是熔點(diǎn)相對(duì)較低,腐蝕性比氟化物小,同時(shí)對(duì)錒系(An)和鑭系(Ln)等的化合物熔解度較高[5],這些混合熔鹽及應(yīng)用的基本性質(zhì)見(jiàn)表1。

表1 各種氯化物混合熔鹽的組成及應(yīng)用
1 LiCl-KCl共晶熔鹽
LiCl-KCl晶熔鹽體系具有電化學(xué)范圍寬、組成穩(wěn)定和核素離子的溶解度高及操作溫度低等特點(diǎn),因此廣泛應(yīng)用于電解精煉和熔鹽還原萃取等后處理工藝。
1.1電解精煉
由美國(guó)阿貢國(guó)家實(shí)驗(yàn)室(Argonne National Laboratory,ANL)發(fā)展的高溫電化學(xué)冶金(即電解精煉)用于處理金屬乏燃料是極具前景的干法后處理技術(shù)[6]。同時(shí),電解精煉由于工藝簡(jiǎn)單和操作溫度較低等優(yōu)點(diǎn)日益受到世界各國(guó)的關(guān)注和重視。國(guó)際上采用電解精煉技術(shù)開(kāi)展乏燃料后處理工藝研究的主要國(guó)家和組織有美國(guó)、歐盟、日本、韓國(guó)和意大利等。金屬乏燃料的電解精煉流程主要包括電解、陰極處理、金屬鑄造和廢鹽處理等工藝。乏燃料電解精煉過(guò)程如圖1所示[7],剪切后的金屬或還原后的乏燃料置于陽(yáng)極籃中。An、堿金屬、堿土金屬和Ln等裂變產(chǎn)物(FPs)元素熔解成為離子:


圖1 乏燃料電解精煉過(guò)程的原理[7]
Ln等在熔鹽中積聚,而活潑金屬元素和大部分Zr則留于陽(yáng)極籃中。由圖2可知,U的還原電位與其他An和Ln相差較大,可選擇性地沉積于固體陰極上;而在液鎘陰極(LCC)上An與Ln可組分離,即剩余的U和Pu與次錒系(Minor Actinide,MA)元素回收于液態(tài)金屬陰極[8]中:


圖2 450℃下LiCl-KCl熔鹽中不同的Ln和An于不同的工作電極上的表觀電勢(shì)[8]
1.1.1美國(guó)
美國(guó)是最早開(kāi)始高溫熔鹽電解處理乏燃料的國(guó)家之一,主要研究機(jī)構(gòu)有阿貢國(guó)家實(shí)驗(yàn)室和愛(ài)達(dá)荷國(guó)家實(shí)驗(yàn)室(Idaho National Laboratory,INL)等。ANL研發(fā)熔鹽電解后處理流程主要是為了處理金屬乏燃料,并從中回收U和Pu,并以此為基礎(chǔ)擴(kuò)展至金屬氧化物、碳化物及其他乏燃料的干法后處理領(lǐng)域。ANL在干法后處理中取得重大的突破,包括[9]:鈾的電解精煉、鈾金屬的純化、研發(fā)了Mark-Ⅳ和Ⅴ型電解精煉裝置(見(jiàn)圖3)、采用電化學(xué)還原法從輕水反應(yīng)堆(LWR)燃料中回收鈾和其他An元素用于快堆的燃料制造,實(shí)現(xiàn)了核燃料的閉式循環(huán)。ER涉及的主要問(wèn)題包括電流效率、U的回收率、Zr的回收和Cd池系統(tǒng)與其他元素的相互作用等。

圖3 ANL燃料處理設(shè)施中Mark-Ⅳ和(a)和Mark-Ⅴ(b)型電解精煉裝置[9]
ANL正在研究干法后處理流程的改進(jìn),以提高電解精煉效率并擴(kuò)大規(guī)模,主要的發(fā)展方向:1)增大電解精煉裝置的通量,并使其適用于LWR的乏燃料的處理;2)實(shí)現(xiàn)產(chǎn)物回收的自動(dòng)化以增大通量;3)發(fā)展陰極間歇性去除An以提高效率;4)評(píng)估并測(cè)試電解精煉裝置模塊的設(shè)計(jì),使其適用于商業(yè)化生產(chǎn)[10]。
INL采用干法處理乏燃料的時(shí)間悠久,從1964 年9月至1969年,INL處理了約5t的燃料(約35000根金屬燃料棒),同時(shí)包括燃料的遠(yuǎn)程制造并用于快堆。INL從1996年6月開(kāi)始采用干法處理實(shí)驗(yàn)增殖堆(Experimental Breeder Reactor-Ⅱ,EBR-Ⅱ)的金屬乏燃料,至2007年5月已處理了約830 kg EBR-Ⅱ的驅(qū)動(dòng)燃料和2 600 kg包層燃料[11]。INL的EBR-Ⅱ的燃料處理設(shè)施(Fuel conditioning facility,F(xiàn)CF)的實(shí)驗(yàn)表明該技術(shù)可用于乏燃料的后處理,需驗(yàn)證的過(guò)程包括乏燃料的分離、U-Pu-Zr合金燃料的制備和高放廢棄物的處理等[11]。
1.1.2ITU
歐盟(EU)進(jìn)行高溫熔鹽電解后處理的機(jī)構(gòu)為聯(lián)合合作中心的超鈾元素研究所(Joint Research Centre-InstituteforTransUraniumElements,JRC-ITU)。除了研究惰性電極于熔鹽中用于電解精煉外,ITU 于2002年發(fā)展基于固體鋁電極的創(chuàng)新性An系元素整體回收法用于乏燃料后處理。該回收法主要步驟有電解精煉、全電解和氯化分離等,具體步驟見(jiàn)圖4。

圖4 ITU提出的采用固體Al陰極的電解精煉后處理流程[7]
An與Ln分離的關(guān)鍵是最不活潑的An系元素與最活潑的Ln系元素的表觀標(biāo)準(zhǔn)電勢(shì)差[7]。由圖2可知,活潑Al電極上兩者的電勢(shì)差可達(dá)到150 mV,而液體Cd和固體W電極上兩者的電勢(shì)差僅為75 mV,因此固體Al電極適合用于An系元素的整體回收。同時(shí),固體Al電極上An與Al主要生成An-Al3和An-Al4等合金化合物,An在每1 g Al上平均沉積量約為1.9 g[7]。ITU通過(guò)采用未輻照過(guò)METAPHIX燃料(U61-Pu22-Am2-Gd0.5-Y0.5-Ce0.5-Zr10)進(jìn)行實(shí)驗(yàn),結(jié)果表明在Al電極上得到一層均一致密的沉積層,其中An元素的質(zhì)量分?jǐn)?shù)大于99.9%[12]。全電解的主要目的是電解回收An后的熔鹽凈化回收,而氯化分離則是將An與Al通過(guò)二者氯化物蒸汽壓的差異進(jìn)行分離的關(guān)鍵步驟。
1.1.3日本
目前,日本采用干法后處理乏燃料的主要研究機(jī)構(gòu)有電力工業(yè)中央研究所(Central Research Institute of Electric Power Industry,CRIEPI)和日本原子能機(jī)構(gòu)(Japan Atomic Energy Agency,JAEA)。
CRIEPI從20世紀(jì)80年代開(kāi)始研究干法后處理技術(shù),主要工作是進(jìn)行快堆核燃料循環(huán)的可行性研究,重點(diǎn)研究干法后處理技術(shù),包括:金屬燃料及快增殖堆的循環(huán)、PUREX流程產(chǎn)生的HLLW的干法分離超鈾元素(TRU)和LWR金屬氧化物燃料的化學(xué)/電化學(xué)還原及干法后處理[13]。
由于氮化物燃料具有較高的中子能譜寬度和導(dǎo)熱性,因此適合作為MA嬗變材料。JAEA于20世紀(jì)90年代開(kāi)始在LiCl-KCl熔鹽中進(jìn)行氮化物燃料的電解精煉[14],其流程主要包括氮化物燃料的電解精煉和An再氮化2個(gè)過(guò)程。
1.1.4韓國(guó)
韓國(guó)干法后處理技術(shù)的主要研究機(jī)構(gòu)為韓國(guó)原子研究所(Korea Atomic energy Research Institute,KAERI)。KAERI從1997年開(kāi)始進(jìn)行干法后處理乏燃料技術(shù)的研究[15],主要包括預(yù)處理、氧化物還原、電解精煉和電解萃取及廢鹽處理等工藝。KAERI研制了用于高通量的電解精煉裝置(HTER)、熔鹽蒸餾裝置和工程級(jí)鑄造熔爐[15]等設(shè)備。KAERI已與PRIDE工廠合作設(shè)計(jì)出了可年產(chǎn)10 t鈾的HTER;實(shí)驗(yàn)室級(jí)的熔鹽蒸餾裝置,已具有每年批次處理20 kg U的能力[15]。
發(fā)展HTER是實(shí)現(xiàn)韓國(guó)實(shí)行乏燃料經(jīng)濟(jì)上可行的關(guān)鍵之一。KAERI設(shè)計(jì)HTER時(shí),引入了創(chuàng)新性想法[15]:1)無(wú)Cd池;2)單獨(dú)裝置中制備UCl3;3)采用自行剝落的石墨陰極;4)采用半連續(xù)裝置回收沉積物。
1.2熔鹽還原萃取
高溫熔鹽還原萃取為還原劑存在條件下,采用液體金屬萃取實(shí)現(xiàn)氟鹽中An的分離和氯化物熔鹽中次錒系元素的分離[2]。對(duì)于熔鹽堆(Molten Salt Reactor,MSR),后處理的目的是萃取FPs元素,使錒系元素(An)留于熔鹽中并在反應(yīng)堆中循環(huán)使用;相反,對(duì)于固體核燃料,干法后處理的目的是為了萃取Ans,而使FPs留于熔鹽中[10]。
還原萃取主要有低溫氯化物L(fēng)iCl-KCl/A和高溫氟鹽LiF-AlF3/Al這2個(gè)體系[10],其中采用液體金屬A進(jìn)行的還原萃取分離An/FPs的原理[16]:

其中,MCln代表鹽相中的氯化物。采用LiCl-KCl/Cd 或Bi體系還原萃取分離錒系元素和Ce的研究[16]表明,773 K時(shí)采用Ga、Bi和Cd,U對(duì)Ce的分離因子分別為3.0×104、846和77,表明液Ga萃取的選擇性較Bi和Cd高。
2 NaCl-2CsCl熔鹽
NaCl-2CsCl可作為處理氧化物和MOX乏燃料的電化學(xué)沉積熔鹽介質(zhì)[17]。俄羅斯于20世紀(jì)70年代開(kāi)始進(jìn)行FREGAT項(xiàng)目;20世紀(jì)80年代俄羅斯核反應(yīng)堆研究所(RIAR)建立了BN-600燃料的電化學(xué)生產(chǎn)工藝,每季度可產(chǎn)1 500 kg MOX燃料;從20世紀(jì)90年代開(kāi)始,RIAR與美國(guó)能源部(DOE)和CEA合作進(jìn)行電化學(xué)法處理武器級(jí)Pu的研究[2]。
氧化物乏燃料后處理中,U和Pu的以下特殊性質(zhì)是十分重要[18]:1)從電化學(xué)角度上,U和Pu的氧化物在熔鹽中熔解或陽(yáng)極氧化時(shí),會(huì)形成MeO2n+絡(luò)合物,并可在陰極還原為相應(yīng)的氧化物;2)高溫(T>400℃)條件下,UO2和PuO2可導(dǎo)電;3)氯化物熔鹽中,U的穩(wěn)定價(jià)態(tài)為U(Ⅲ)、U(Ⅳ)和U(Ⅵ),而Pu的氧化態(tài)只在特定電勢(shì)下存在,熔鹽中通入Cl2和O2混合氣體方可得到所需濃度的PuO22+;4)無(wú)論P(yáng)u起始處于何種氧化態(tài),通過(guò)改變條件可轉(zhuǎn)化為氧化物形式,而在氧化條件下,U溶解于熔鹽中;5)U和Pu的氧化物于較正電位還原,而大部分FPs于較負(fù)電位還原,因此電沉積UO2和PuO2過(guò)程中可實(shí)現(xiàn)FPs的分離。
基于UO2和PuO2和FPs在熔鹽中特殊性質(zhì),RIAR發(fā)展了Dimitrovgrad干法流程(Dimitrovgrad Dry Process,DDP)。DDP法的主要技術(shù)[18]:1)UO2的電沉積技術(shù);2)PuO2的沉淀結(jié)晶技術(shù);3)UO2和PuO2的共沉積。
RIAR熔鹽干法后處理方向:從鈾基乏燃料中回收UO2、從MOX燃料中回收Pu和由MOX乏燃料后處理后制造MOX燃料。DDP法用于氧化物燃料和MOX乏燃料處理,具體過(guò)程[2]:1)NaCl-KCl或LiCl-NaCl-KCl-CsCl熔鹽中完成燃料的熔解;2)采用電解法將UO2和Pu分離,U與Pu的分離系數(shù)約為120~140;3)除去FPs得到PuO2沉淀晶體,底部PuO2中Pu的回收率為99.5~99.9%;4)電解除去鈾,大部分FPs可與UO2同時(shí)沉積于陰極;5)采用磷酸鹽沉淀鑭系(Ln)和次錒系(MA)元素以熔鹽凈化。采用DDP法處理乏燃料對(duì)主要的FPs的DF值如表2所示,表明采用DDP法可去除大部分裂片離子,達(dá)到分離純化目的。
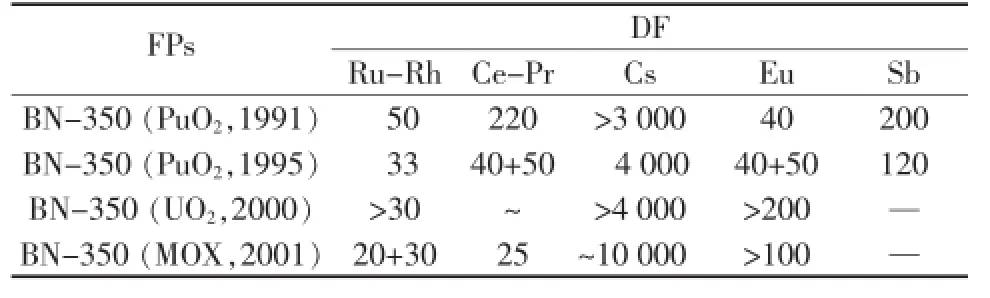
表2 PuO2、UO2和MOX對(duì)主要FPs的去污因子(DF)[2]
3 LiCl-Li2O熔鹽
熔鹽電解精煉前,氧化物或混合氧化物乏燃料需還原為金屬。目前,主要采用電化學(xué)還原法,使用的熔鹽介質(zhì)為L(zhǎng)iCl-Li2O,陽(yáng)極和陰極分別發(fā)生UO2的還原和O2-的氧化過(guò)程。
LiCl-Li2O熔鹽中氧化物乏燃料陰極還原為金屬的電化學(xué)機(jī)理如圖5所示[19]。
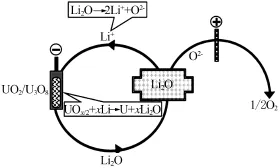
圖5 電化學(xué)還原過(guò)程的原理[19]

產(chǎn)生的O2-擴(kuò)散至惰性金屬電極或碳電極上生成O2或CO及CO2:

通常LiCl熔鹽中錒系元素氧化物的還原率約為95%,但采用揮發(fā)氧化(Voloxidation),將UO2轉(zhuǎn)化為U3O8再用H2還原,可得到疏松多孔結(jié)構(gòu)的乏燃料塊,縮短還原時(shí)間的同時(shí)可將還原率提高至99.6%[20]。
4 總結(jié)與展望
隨著中國(guó)快堆的快速發(fā)展及乏燃料后處理研究的深入,無(wú)機(jī)氯化物熔鹽在其中發(fā)揮著重要的作用。干法后處理中主要應(yīng)用的混合熔鹽有LiCl-KCl、NaCl-2CsCl和LiCl-Li2O體系,分別應(yīng)用于電解精煉及還原萃取、氧化物電沉積和電化學(xué)還原等領(lǐng)域。
目前,中國(guó)熔鹽后處理正處于起步階段,與國(guó)際先進(jìn)水平仍存在一定的差異。中國(guó)可在氯化物熔鹽方面的研究包括:1)高純氯化物熔鹽的制備及后處理過(guò)程中產(chǎn)生的廢鹽凈化設(shè)備和技術(shù)的研制;2)含有錒系氯化物熔鹽性質(zhì)包括熔鹽密度、黏度、蒸汽壓和電導(dǎo)率等性質(zhì)的研究;3)高通量熔鹽電解和陰極處理及傳送等設(shè)備的研制及熔鹽電解技術(shù)等方面加強(qiáng)研究。
[1]Uhlír J,Marecek M.Fluoride volatility method for reprocessing of LWR and FR fuels[J].Journal of Fluorine Chemistry,2009,130(1):89-93.
[2]Organisation for Economic Co-operation and Development.Pyrochemical separations in nuclear applications[R].PARIS:OEDC,2004.
[3]Yoo J H,Seo C S,Kim E H,et al.A conceptual study of pyroprocessing for recovering actinides from spent oxide fuels[J].Nucl.Eng. Technol.,2008,40(7):581-592.
[4]Iizuka,M,Koyama T,Sakamura Y,et al.Development of pyro-processing technology at criepi for carving out the future of nuclear fuel cycle[C]∥Proceedings of GLOBAL 2013:International NuclearFuelCycleConference,2013.LaGrangePark:AmericanNuclear Society,2013:176-185.
[5]Fukasawa K.Systematic study on the thermodynamic stability of lanthanides and actinides in molten alkali and alkaline earth chlorides[D].Kyoto:Kyoto University,2012.
[6]Koyama T,Hijikata T,Usami T,et al.Integrated experiments of electrometallurgical pyroprocessing using plutonium oxide[J].Journal of Nuclear Science and Technology,2007,44(3):382-392.
[7]Iizuka M,Uozumi K,Ogata T,et al.Development of an innovative electrorefiner for high uranium recovery rate from metal fast reactor fuels[J].Journal of Nuclear Science and Technology,2009,46(7):699-716.
[8]GlatzJP,Malmbeck R,Soucek P,et al.Development of pyrochemical separation processes for recovery of actinides from spent nuclear fuel in molten LiCl-KCl[M]∥Molten Salts Chemistry.Amsterdam:Elsevier,2013:541-560.
[9]Westphal B R,Vaden D,Li S X,et al.Fate of noble metals during the pyroprocessing of spent nuclear fuel[C]∥Proceedings of Global,2009:Paris,2009.
[10]Argonne National Laboratory.Pyroprocessing technologies:recyc
ling used nuclear fuel for a sustainable energy future[Z/OL]. Argonne National Laboratory,2012-06-04[2015-10-22].http:∥www.ne.anl.gov/pdfs/12_Pyroprocessing_bro_5_12_v14[6].pdf.
[11]Benedict R W,Solbrig C,Westphal B,et al.Pyroprocessing progress at idaho national laboratory[C]∥Global,2007.Idaho Falls:Idaho National Laboratory,2007.
[12]Conocar O,Douyere N,Glatz J P,et al.Promising pyrochemical actinide/lanthanide separation processes using aluminum[J].Nuclear Science and Engineering,2006,153(3):253-261.
[13]Murakami T,Uozumi K,Sakamura Y,et al.Recent achievements and remaining challenges on pyrochemical reprocessing in CRIEPI[C]∥ProceedingsoftheFirstACSEPTInternationalWorkshop:Portugal,2010.
[14]Arai Y,Akabori M,Minato K.JAEA′s activities on nitride fuel researchforMAtransmutation[C]∥Proc.9th IEMPT:N?mes,F(xiàn)rance,2006.
[15]Kim J G.Li S J,Woo M S,et al.Recent advances in electrorefining process in KAERI[C]∥Proceedings of GLOBAL 2011:Makuhari,Japan,2011.
[16]TodaT,Maruyama T,Moritani K,et al.Thermodynamic properties oflanthanidesandactinidesforreductiveextractionofminoractinides[J].Journal of Nuclear Science and Technology,2009,46(1):18-25.
[17]Uehara A,Nagai T,F(xiàn)ujii T,et al.Spectrophotometric and electrochemicalstudyofneptuniumionsinmoltenNaCl-CsCleutectic[J]. Journal of Nuclear Materials,2013,437(1/2/3):166-170.
[18]Vavilov S,Kobayashi T,Myochin M.Principle and test experience of the RIAR′s oxide pyro-process[J].Journal of Nuclear Science and Technology,2004,41(10):1018-1025.
[19]Hur J M,Jeong S M,Lee H.Underpotential deposition of Li in a molten LiCl-Li2O electrolyte for the electrochemical reduction of U from uranium oxides[J].Electrochemistry Communications,2010,12(5):706-709.
[20]Choi E Y,Lee J W,Park J J,et al.Electrochemical reduction behavior of a highly porous SIMFUEL particle in a LiCl molten salt[J].Chemical Engineering Journal,2012,207/208:514-520.
聯(lián)系方式:yeguoan@ciae.ac.cn
Application progress of inorganic molten chlorides in dry reprocessing of spent fuel
Wang Youqun,He Hui,Lin Rushan,Ye Guoan,Tang Hongbin,Jia Yanhong
(Department of Radiochemistry,China Institute of Atomic Energy,Beijing 102413,China)
The application progress of inorganic molten chlorides in the spent fuel reprocessing was reviewed.LiCl-KCl eutectic used in the electrorefining and reductive extraction of metal spent fuel,and molten salt of LiCl-Li2O utilized in the reduction of oxide or mixed oxide(MOX)spent fuel,and NaCl-2CsCl mixture applied in the electrowinning were mainly introduced.Finally,the application future and development direction of molten chlorides in the spent fuel dry reprocessing of China was prospected.
chlorides molten salt;spent nuclear fuel;dry reprocessing
TQ124.4
A
1006-4990(2016)08-0001-05
國(guó)家自然科學(xué)基金(9122620)。
2016-02-22
王有群(1988—),男,博士研究生,主要研究方向?yàn)楦煞ㄌ幚恚压_(kāi)發(fā)表文章6篇。
葉國(guó)安
猜你喜歡
汽車實(shí)用技術(shù)(2022年15期)2022-08-19 02:48:28
今日農(nóng)業(yè)(2020年20期)2020-12-15 15:53:19
中華養(yǎng)生保健(2020年7期)2020-11-16 01:13:34
能源(2018年10期)2018-12-08 08:02:48
電子制作(2018年16期)2018-09-26 03:27:00
石油煉制與化工(2017年5期)2017-04-06 19:47:30
能源(2016年10期)2016-02-28 11:33:30
汽車實(shí)用技術(shù)(2015年8期)2015-12-26 09:01:02
中國(guó)慣性技術(shù)學(xué)報(bào)(2015年1期)2015-12-19 13:12:05
汽車維修與保養(yǎng)(2015年2期)2015-04-17 01:30:39
