HPLC法同時測定馬兜鈴中4種成分
2016-04-06 01:23:44田禾苗程雪梅王長虹王崢濤上海中醫藥大學中藥研究所中藥標準化教育部重點實驗室中藥新資源與質量標準綜合評價國家中醫藥管理局重點實驗室上海2020上海中藥標準化研究中心上海2020
中成藥 2016年3期
關鍵詞:中藥
田禾苗, 程雪梅,2, 王長虹,2*, 王崢濤,2(.上海中醫藥大學中藥研究所,中藥標準化教育部重點實驗室,中藥新資源與質量標準綜合評價國家中醫藥管理局重點實驗室,上海2020;2.上海中藥標準化研究中心,上海2020)
?
HPLC法同時測定馬兜鈴中4種成分
田禾苗1, 程雪梅1,2, 王長虹1,2*, 王崢濤1,2
(1.上海中醫藥大學中藥研究所,中藥標準化教育部重點實驗室,中藥新資源與質量標準綜合評價國家中醫藥管理局重點實驗室,上海201210;2.上海中藥標準化研究中心,上海201210)
摘要:目的 建立同時測定馬兜鈴Aristolochia debilis Sieb.et Zucc.中馬兜鈴酸III、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ的HPLC方法。方法 馬兜鈴70%甲醇提取液的分析采用Agi1ent Extend C18色譜柱(4.6 mm× 250 mm,5 μm);流動相為乙腈(A)-0.1%甲酸水(B),梯度洗脫;體積流量1.0 mL/min;柱溫30℃,檢測波長254 nm。結合大鼠口服馬兜鈴酸I的半數致死量(LD50)及細辛藥材中馬兜鈴酸I的限量,確定馬兜鈴中馬兜鈴酸的限量。結果 馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ分別在0.34~31.90、0.32~30.20、0.12~28.10和0.21~41.30 μg/mL范圍內線性關系良好。馬兜鈴中馬兜鈴酸Ⅰ或馬兜鈴總酸的限量為0.000 3%。結論 該方法準確可靠,可用于馬兜鈴中馬兜鈴酸的限量測定。
關鍵詞:馬兜鈴;馬兜鈴酸Ⅲ;7-羥基馬兜鈴酸Ⅰ;馬兜鈴內酰胺Ⅰ;馬兜鈴酸Ⅰ;LD50;限量;HPLC
Aristo1ochic acidⅢ,7-hydroxy aristo1ochic acidⅠ,aristo1ochia 1actamⅠand aristo1ochic acidⅠexhibited good 1inear re1ationships within the ranges of 0.34 -31.90 μg/mL,0.32 -30.20 μg/mL,0.12 -28.10 μg/mL and 0.21 -41.30 μg/mL,respective1y.The dose 1imitation of aristo1ochic acidⅠor tota1aristo1ochic acids in A.debilis was determined to be nomore than 0.000 3%.C0 NCLUSI0 N This accurate and stab1emethod can be used for the 1imitation determination of aristo1ochic acid in A.debilis.
KEY W 0RDS:Aristolochia debilis Sieb.et Zucc;aristo1ochic acidⅢ;7-hydroxy-aristo1ochic acidⅠ;aristo1ochia 1actamⅠ;aristo1ochic acidⅠ;LD50;dose 1imitation;HPLC
馬兜鈴為馬兜鈴科植物北馬兜鈴Aristolochia debilis Sieb.et Zucc.的干燥成熟果實,具有清肺降氣、止咳平喘、清腸消痔之功效[1],其化學成分有馬兜鈴酸、生物堿、木脂素、甾體、萜類等,以馬兜鈴酸類成分為主。同時,馬兜鈴酸類成分又可分為馬兜鈴酸和馬兜鈴內酰胺兩大類[2],前者主要有馬兜鈴酸Ⅰ、Ⅱ、Ⅲ等,而后者主要為馬兜鈴內酰胺Ⅰ、Ⅱ、Ⅲa等。現代研究表明,馬兜鈴具有抗腫瘤、抗菌、抗炎、鎮痛等藥理作用[3],但長期服用含馬兜鈴酸的藥物可能造成慢性腎衰竭以及腎小管病變。其機制是與DNA形成加合物,馬兜鈴酸在體內經簡單代謝過程后,其胺離子將被硝基還原酶還原為環內酰胺離子[4],一部分代謝產物被還原為馬兜鈴內酰胺,進入腎小管上皮細胞、蓄積于細胞質內發揮毒性作用[5],而另一部分則與DNA中嘌呤核苷酸的環外氨基共價結合,形成特異性AA-DNA加合物[6],進而使IAS基因和p53基因發生突變。
近年來,長期服用含馬兜鈴酸的藥物所造成的副作用已引起國際社會的高度關注,并冠以“中草藥腎病”,對中醫藥的健康發展產生了嚴重的影響。1999年,英國藥物安全委員會(CSM)建議應立即禁止使用含有馬兜鈴的中草藥,同時英國醫藥管理局(MCA)也提交了一項關于禁止可能含有馬兜鈴的中草藥在英國進口、銷售和供應的禁令,并進行表決,結果自1999年7月至10月在全英范圍內暫時性禁用相關產品,但不久就延續為無限期禁用[7]。因此,《中國藥典》從2005年版起,已不再收錄關木通、廣防己、青木香等含有馬兜鈴的中藥方,并以其他品種替代,而且對其他含馬兜鈴酸生藥的藥用部位也進行了更改。例如,2000年版以前的《中國藥典》中細辛藥用部位均為“全草”,但后來考慮到其地上部分馬兜鈴酸含有量較高,而且古代中醫一直僅使用根部,《日本藥局方》也僅用“根和根莖”,所以從2005年起《中國藥典》亦將藥用部位修訂為根和根莖;《中國藥典》2010年版進一步規定,依照HPLC法測定,按干燥品計算細辛中馬兜鈴酸Ⅰ的含有量不得過0.001%[8],但相關鑒別項下只對馬兜鈴酸Ⅰ進行薄層鑒別[1],而有關馬兜鈴中馬兜鈴酸類成分的限量尚無規定,為其安全使用帶來了一定的安全隱患。為了嚴格控制馬兜鈴藥材質量,保證用藥安全,本實驗建立了同時測定馬兜鈴中馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ含有量的HPLC法,并根據馬兜鈴酸I的相關毒性數據(LD50)推定限量,為其安全標準的制定奠定基礎。
1 儀器與試藥
Agi1ent 1100高效液相色譜儀,包括Agi1ent ChemStation中文色譜工作站;KQ-250DB數控超聲波清洗儀;Mett1er 200電子天平;Agi1ent Extend C18色譜柱(4.6 mm×250 mm,5 μm)。馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ對照品(上海盛中醫藥化工有限公司,純度均>95%);馬兜鈴內酰胺Ⅰ(天津士蘭科技有限公司,純度>98%);馬兜鈴酸Ⅰ對照品(上海中藥標準化研究中心,純度≥98%)。乙腈、甲酸為色譜純;水為超純水;其他試劑為分析純。15批馬兜鈴藥材購自黑龍江、遼寧、新疆、湖南、陜西、四川等多個產區的藥房及中藥材市場,由上海中藥標準化研究中心吳立宏研究員鑒定,其性狀均與《中國藥典》2010年版規定一致,故為正品。
2 方法與結果
2.1 色譜條件 Agi1ent Extend C18色譜柱(4.6 mm×250 mm,5 μm);流動相為乙腈(A)-0.1%甲酸水(B),梯度洗脫(0~35 min,24%~30%A;35~50 min,30%~55%A;50~60 min,55%A);體積流量1.0 mL/min;柱溫30℃;檢測波長254 nm。在該色譜條件下,各檢測組分的分離度良好,見圖1。
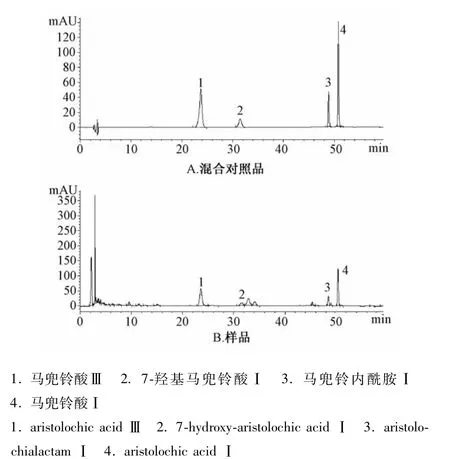
圖1 HPLC色譜圖Fig.1 HPLC chromatograms
2.2 對照品溶液的制備 馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ分別精密稱取馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ適量,加甲醇制成每1 mL含0.16 mg馬兜鈴酸Ⅲ、0.15 mg 7-羥基馬兜鈴酸Ⅰ、0.14 mg馬兜鈴內酰胺Ⅰ、0.20 mg馬兜鈴酸Ⅰ的溶液,即得。
2.3 供試品溶液的制備 精密取本品細粉(過四號篩)0.5 g,置于具塞錐形瓶中,精密加入70%甲醇25 mL,密塞,稱定重量,超聲(250 W、40 kHz)60 min,放冷,再稱定重量,70%甲醇補足減失的重量,搖勻,濾過,精密量取續濾液2 mL,置于10 mL量瓶中,加70%甲醇至刻度,搖勻,濾過,取續濾液,即得。
2.4 含有量測定方法 分別吸取對照品與供試品溶液20 μL,注入HPLC色譜儀,在“2.1”項條件下,外標一點法測定馬兜鈴酸Ⅲ、7-OH馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ的含有量。
2.5 線性關系的考察 精密吸取“2.2”項下對照品溶液,配置成含31.90、25.50、19.10、12.70、6.40、3.20、0.32 μg/mL馬兜鈴酸Ⅲ,30.20、24.20、18.10、12.10、6.00、3.00、0.30 μg/mL 7-羥基馬兜鈴酸I,28.10、22.40、16.80、11.20、5.62、0.30、0.10 μg/mL馬兜鈴內酰胺Ⅰ和41.30、33.10、24.80、16.50、8.25、2.08、0.21 μg/mL馬兜鈴酸Ⅰ的混合對照品溶液,進樣20 μL分析。結果表明,各成分在相應的范圍內線性關系良好,具體見表1。

表1 4種成分的線性關系和范圍Tab.1 Linear relationships and ranges of four consitutents
2.6 精密度試驗 精密吸取高、中、低3種不同質量濃度的對照品溶液各20 μL,連續進樣6次,分別計算馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ和馬兜鈴酸Ⅰ的日內、日間精密度。結果,4種成分峰面積的日內、日間精密度ISD均≤3%,表明儀器精密度良好,具體見表2。
2.7 重復性試驗 精密稱取1號樣品細粉0.5 g,按“2.3”項下方法制備供試品溶液,平行6份,測得馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ和馬兜鈴酸Ⅰ的平均含有量分別為0.13%、 0.02%、0.02%、0.08%,ISD分別為0.81%、1.9%、1.9%、0.90%,表明該方法重復性良好。

表2 4種成分的日內、日間精密度Tab.2 Intra-day and inter-day precisions of four consitutents
2.8 穩定性試驗 精密吸取供試品溶液20 μL,于0、1、3、8、12、18、24、36 h進樣,測定馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ和馬兜鈴酸I峰面積的ISD分別為0.59%、0.87%、2.7%、2.0%,表明供試品溶液在36 h內穩定。
2.9 加樣回收率試驗 精密稱取含有量已知的馬兜鈴藥材(1號)9份,每份0.25 g,分別按含有量的80%、100%、120%加入對照品溶液,平行3份,按“2.3”項下方法制備供試品溶液,計算加樣回收率,結果見表3。
2.10 含有量測定結果 見表4。
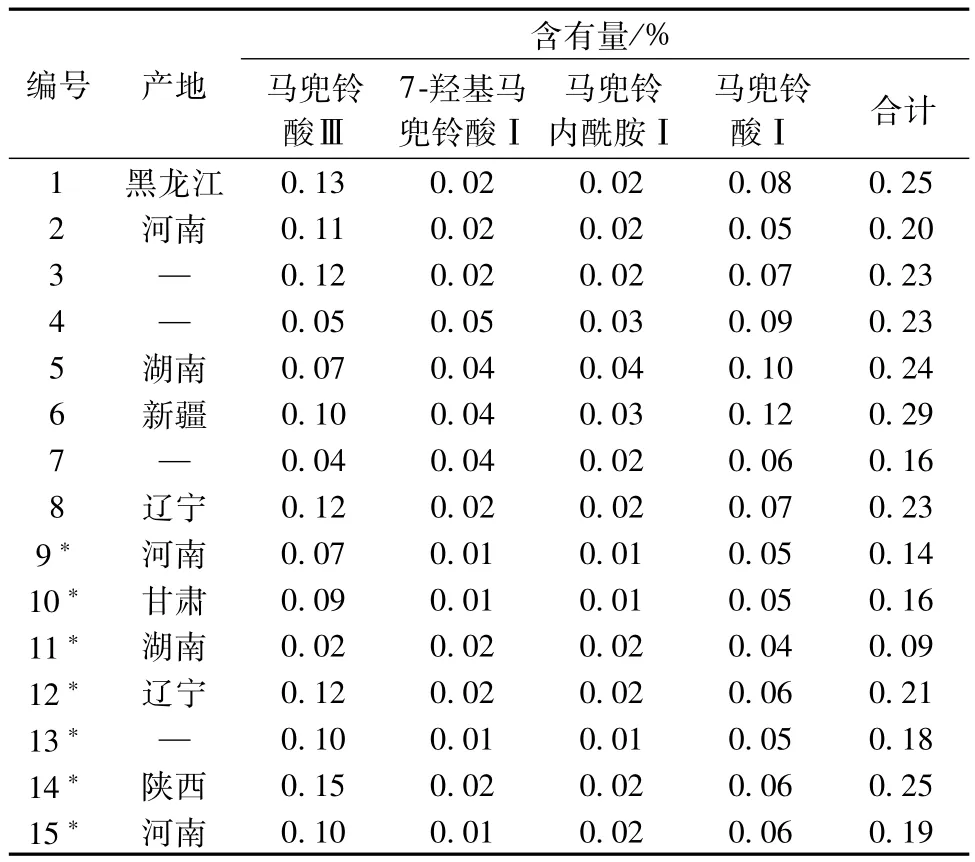
表4 4種成分含有量的測定結果Tab.4 Determ ination results of the contents of four constituents

表3 加樣回收率試驗結果Tab.3 Results of recovery tests
2.11 馬兜鈴酸的限量研究 查閱文獻[9],得到了NMI I小鼠(美國馬里蘭貝賽斯達鼠)和Wistar大鼠在灌胃給予馬兜鈴酸(77.24%馬兜鈴酸Ⅰ和21.18%馬兜鈴酸Ⅱ鈉鹽的混和溶液)后的LD50,見表5。
由表可知,不同種屬動物服用馬兜鈴酸后,其LD50差別較大,由Wistar大鼠轉化后的含有量上限約為NMI I小鼠的4倍。但為了嚴格控制馬兜鈴中總酸的含有量,本實驗采取了由NMI I雄性小鼠轉化后的含有量上限作為馬兜鈴中馬兜鈴酸Ⅰ的含有量上限(雄性NMI I小鼠口服馬兜鈴酸的LD50為55.9 mg/kg,LD5為LD50的1/19,即2.94 mg/kg。人與小鼠的劑量換算比例為1∶9.1,即LD5換算成人的致毒劑量為0.32 mg/kg)。以人LD5劑量的1/10為安全劑量上限,按平均體重60 kg計算,并以馬兜鈴藥材最大服用量9 g計,計算出馬兜鈴藥材中馬兜鈴酸Ⅰ的含有量不應超過0.02%。
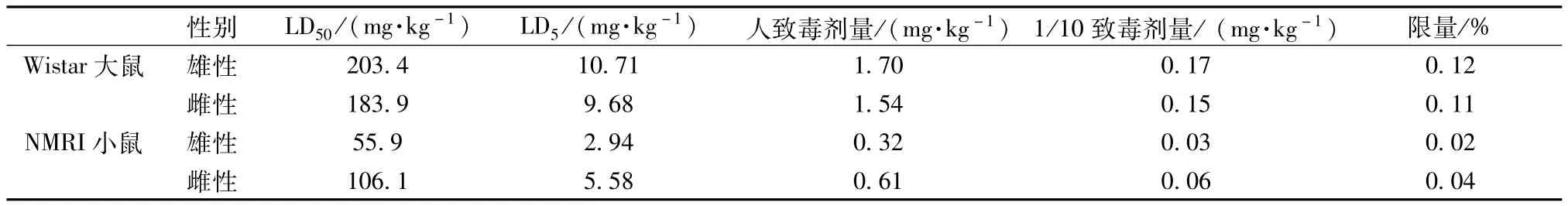
表5 NMRI小鼠和W istar大鼠的LD50及限量Tab.5 LD50and dose lim itations of NMRIm ice and W istar rats
同時,還測定了馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ的含有量,但由于現有文獻并未這3種成分的LD50進行報道,故無法對其的含有量上限做出精確限定。據報道,其他馬兜鈴酸類化合物,如馬兜鈴酸Ⅱ、馬兜鈴酸Ⅲa、馬兜鈴酸ⅠVa等毒性和馬兜鈴酸Ⅰ相比,可能更大[10]。因此,用馬兜鈴酸Ⅰ的毒性代替其它3種馬兜鈴酸類成分具有一定的合理性,即采用4種馬兜鈴酸的總和進行限量控制是可行的。因此,暫定馬兜鈴藥材及飲片中馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ的總量不得超過0.02%。
細辛也為含馬兜鈴酸類成分的藥材,《中國藥典》2010年版規定,其中馬兜鈴酸Ⅰ的含有量不得超過0.001%[8]。因此,細辛和馬兜鈴的用量分別為1~3 g和3~9 g,故推算馬兜鈴藥材或飲片中馬兜鈴酸I或馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ的總量均不得超過0.000 3%。
3 討論
3.1 指標成分的選擇 馬兜鈴酸類成分是馬兜鈴藥材中的主要有毒成分,又包括馬兜鈴酸和馬兜鈴內酰胺兩大類。其中,馬兜鈴酸Ⅰ是最常見的馬兜鈴酸類化合物,存在于幾乎所有的馬兜鈴屬植物中,而馬兜鈴內酰胺Ⅰ作為次生代謝產物,普遍存在于馬兜鈴屬各科植物中[11]。因此,以馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ為指標成分,能更加全面地評價和控制馬兜鈴的毒性。3.2 檢測波長的選擇 采用二極管陣列檢測器,在190~400 nm波長下對供試品和對照品溶液中的成分進行全波長掃描。結果,馬兜鈴酸Ⅲ的最大吸收波長在254 nm下為強吸收;7-羥基馬兜鈴酸Ⅰ在215 nm和275 nm下都有很強的紫外吸收,并且在300~320 nm處為強吸收肩峰;馬兜鈴內酰胺Ⅰ在235、254、290 nm下都有強吸收;馬兜鈴酸Ⅰ的最大吸收峰在225、254、330 nm處。因此,為了同時測定這4種馬兜鈴酸類成分,選擇254 nm波長處為佳。3.3 供試品溶液制備方法的選擇 本實驗分別考察了不同比例甲醇和純水對馬兜鈴藥材中馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ和馬兜鈴酸Ⅰ提取效率的影響。結果表明,以70%甲醇提取時,4種成分的提取效率均最高。同時,還比較了超聲提取和甲醇回流提取對其提取率的影響,但發現兩種方法下馬兜鈴內酰胺Ⅰ的峰面積均逐漸增加,可能是因為其他不穩定的馬兜鈴酸類成分受熱轉化為馬兜鈴內酰胺Ⅰ,但超聲提取增加的幅度小于回流提取。因此,為了確保馬兜鈴內酰胺Ⅰ的準確檢測,最終選擇超聲提取60 min。
3.4 馬兜鈴酸限度的選擇 鑒于馬兜鈴藥材的毒性,根據其用量和文獻報道馬兜鈴酸Ⅰ的相關毒性數據(LD50)及細辛藥材中馬兜鈴酸Ⅰ的限量,推定馬兜鈴藥材中馬兜鈴酸的限量,為其安全標準的制定奠定基礎。按照小鼠口服馬兜鈴酸Ⅰ后的LD50和藥典規定的馬兜鈴藥材最大劑量,推算出馬兜鈴藥材及飲片中馬兜鈴酸Ⅲ、7-羥基馬兜鈴酸Ⅰ、馬兜鈴內酰胺Ⅰ、馬兜鈴酸Ⅰ的總含有量均不得超過0.02%。但是,如果按照同為含馬兜鈴酸類成分的細辛中馬兜鈴酸I的限量為0.001%[8]計算,馬兜鈴藥材和飲片中該類成分的限量應定不得超過0.000 3%。本實驗所有樣品中總馬兜鈴酸類成分或馬兜鈴酸Ⅰ的含有量均超過0.02%或0.000 3%,故建議《中國藥典》對馬兜鈴藥材制定合理的限度標準。
參考文獻:
[1] 國家藥典委員會.中華人民共和國藥典:2010年版一部[S].北京:中國醫藥科技出版社,2010:48-49.
[2] 王 瑛,潘競先,賈忠建,等.馬兜鈴屬植物化學成分及生物活性研究進展[J].天然產物與開發,2000,12(6):84-93.
[3] 陳孟蘭,朱正蘭.馬兜鈴屬植物的藥理作用研究進展[J].武漢生物工程學院學報,2007,3(1):59-62.
[4] 常菲菲,王友群.馬兜鈴酸體內毒性的產生及防治研究進展[J].藥學進展,2010,34(3):117-124.
[5] 商 樸,王 璇,李曉玫,等.馬兜鈴內酰胺-Ⅰ進入人腎小管上皮細胞及細胞內分布和蓄積的觀察[J].中國中藥雜志,2008,33(7):793-797.
[6] Stiborová M,Frei E,Ar1t V M,et al.Metabo1ic activation of carcinogenic aristo1ochic acid,a risk factor for Ba1kan endemic nephropathy[J].Mutat Res,2008,658(1-2):55-67.
[7] 張子伯,蔣文躍,蔡少青.馬兜鈴酸所致中草藥腎病的醫學和藥學進展及其引發的思索[J].中草藥,2003,34(2):185-188.
[8] 國家藥典委員會.中華人民共和國藥典:2010年版一部[S].北京:中國醫藥科技出版社,2010:214.
[9] Mengs U.Acute toxicity of aristo1ochic acid in rodents[J].Arch Toχicol,1987,59(5):328-331.
[10] 郭永超,林哲絢,李 慧,等.三種馬兜鈴酸類化合物對HK_2
細胞的毒性比較[J].癌變·畸變·突變,2006,2(6):88-92.
[11] Wu T S,Damu A,Su C I,et al.Chemica1 constituents and pharmaco1ogy of Aristolochi species[J].Studies in Nat Prod Chem,2005,32:855-1018.
Simultaneous determ ination of four constituents in Aristolochia debilis by HPLC
TIAN He-miao1, CHENG Xue-mei1,2,WANG Chang-hong1,2*, WANG Zheng-tao1,2
(I.Instituteof ChineseMateria Medica,ShanghaiUniversity of Traditional ChineseMedicine;Key Laboratory of theMinistry of Education for Standardization of Chinese Medicine;Key Laboratory of the State Administration of Traditional Chinese Medicine for New Resources and Quality Evaluation of Chinese Medicine;Shanghai2OI2IO,China;2.Shanghai Research and Development Center for Standardization of ChineseMedicine,Shanghai2OI2IO,China)
ABSTRACT:AIM To estab1ish an HPLCmethod for the simu1taneous determination of aristo1ochic acidⅢ,7-hydroxy-aristo1ochic acidⅠ,aristo1ochia 1actamⅠand aristo1ochic acidⅠin Aristolochia debilis Sieb.et Zucc. METH 0 DS The ana1ysis of 70% A.debilis methano1ic extract was performed on Agi1ent Extend C18co1umn (4.6 mm×250 mm,5 μm),with acetonitri1e(A)-0.1% formic acid(B)as themobi1e phase in a gradient e-1ution manner at1.0 mL/min for f1ow rate,30℃for co1umn temperature and 254 nm for detection wave1ength. Then the dose 1imitation of aristo1ochic acid in A.debilis was determined with reference to themedian 1etha1 dose (LD50)of aristo1ochic acid Iora11y administered to rats and its dose 1imitation in Asurum uropeum.RESULTS
*通信作者:王長虹(1964—),男,研究員,博士生導師,研究方向為中藥新制劑與體內過程。Te1:(021)51322511,E-mai1:wchcxm@163.com
作者簡介:田禾苗,男,碩士生,研究方向為中藥新制劑與體內過程。Te1:15221230656,E-mai1:tianhemiao123456@hotmai1.com
基金項目:《中國藥典》2010版標準提高研究項目(508)
收稿日期:2015-10-30
doi:10.3969/j.issn.1001-1528.2016.03.017
中圖分類號:I284.1
文獻標志碼:A
文章編號:1001-1528(2016)03-0560-06
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國現代中藥(2020年4期)2020-06-10 09:56:34
基層中醫藥(2018年6期)2018-08-29 01:20:20
長春中醫藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49