毒熱清顆粒提取工藝優選
2016-02-15 02:44:59施鈞瀚秦會珍弘孟祥樂
中國醫藥科學 2016年21期
關鍵詞:工藝
施鈞瀚秦會珍,2Δ鄭 弘孟祥樂,2▲
1.河南中醫學院第一附屬醫院,河南鄭州 450000;2.河南大學藥學院,河南開封 475004
毒熱清顆粒提取工藝優選
施鈞瀚1秦會珍1,2Δ鄭 弘1孟祥樂1,2▲
1.河南中醫學院第一附屬醫院,河南鄭州 450000;2.河南大學藥學院,河南開封 475004
目的優選毒熱清顆粒的水提工藝條件。方法以黃芩苷,綠原酸為指標,HPLC測定含量,Agilent ZORBAX XDB-C18色譜柱(4.6mm×150mm,5μm),流動相甲醇-0.4%磷酸梯度洗脫,流速1.0mL/min,檢測波長為280nm(黃芩苷)、327nm(綠原酸)。選加水量、提取次數、提取時間為考察因素,采用L9(34)正交試驗優選毒熱清顆粒的水提取工藝。結果最佳水提取工藝為加12倍量水提取2次,每次2h。結論該工藝有效成分提取完全,工藝簡單,適合于工業生產的需要。
毒熱清顆粒;黃芩苷;正交試驗;高效液相色譜法;提取工藝
毒熱清顆粒方是醫院臨床經驗方。該方藥具有清熱解毒、瀉火散結之功效。用于熱毒壅盛證,癥見壯熱口渴,心胸煩熱、口舌生瘡,瘰疬痰核,疔瘡腫毒,小便黃赤等。方中黃芩清熱燥濕,瀉火解毒;金銀花清熱解毒,疏散風熱,連翹、紫花地丁清熱解毒,涼血消腫散結,甘草清熱解毒,調和諸藥。全方共奏清熱解毒、瀉火散結之功。本文對該方的提取工藝進行研究,擬制成醫院制劑。
1 儀器與試藥
Waters e2695高效液相色譜儀;色譜柱:Agilent XDB-C18柱(4.6mm×150mm,5μm);Sartorius CP225D電子天平,科導臺式超聲波清洗器(Hk250型),AP-01P型真空泵(天津奧特賽恩斯儀器有限公司);黃芩苷對照品(批號:110715-201318)、綠原酸對照品(批號:110753-201314)均來源于中國食品藥品檢定研究院,用于流動相試劑均為色譜純,西格瑪公司生產,水為純化水,其它試劑為分析純。飲片來源于安徽普仁藥業有限公司。
2 方法與結果
2.1 提取工藝的選擇
根據處方中藥材的成分參照參照參考文獻[1-2],采用水煎煮提取工藝,用正交試驗對提取工藝進行優選。
2.2 正交實驗設計
根據文獻[3-7],指標成分選取黃芩苷 、綠原酸,采用L9(34)正交表安排實驗,對加水量、提取時間、提取次數進行考察。因素水平表見表1。
2.3 樣品制備
稱取藥材9份(按處方比例),按表 2試驗分別提取,合并提取液,冷卻(室溫)后測量體積,備用。

表1 因素水平表
2.4 含量測定
2.4.1 對照品溶液的制備 精密稱取黃芩苷對照品11.92mg,加甲醇定容至50mL,制成每1mL含238.2μg的對照品溶液。取綠原酸對照品10.54mg,精密稱定,加甲醇定容至50mL,制成每1mL含210.8μg的對照品溶液。
2.4.2 供試品溶液的制備 量取正交試驗的樣品10.0mL甲醇稀釋至50mL,搖勻靜置,取上清液即得。
2.4.3 陰性對照溶液的制備 缺黃芩樣品溶液:稱取本處方中的藥材(按處方比例不含黃芩),按正交5號的條件制備缺黃芩樣品,然后按供試品溶液制備方法制備,得缺黃芩陰性對照溶液。
缺金銀花樣品溶液:稱取本處方中的藥材(按處方比例不含金銀花),按正交5號的條件制備缺金銀花樣品,然后按供試品溶液制備方法制備,得缺金銀花陰性對照溶液。
2.4.4 色譜條件色譜柱 Agilent XDB-C18柱(4.6mm×150mm,5μm);流動相甲醇-0.4%磷酸梯度洗脫:0~25,甲醇25~47,25~35,甲醇47~47,35~36甲醇47~25,36~40甲醇25~25;檢測波長為280nm(黃芩苷)和327nm(綠原酸)[8-9],流速:1.0mL/min,柱溫25℃。
2.4.5 專屬性試驗 取以上各種樣品溶液(對照品、供試品、陰性溶液)各5μL,依法測定,記錄色譜圖,見圖1。結果供試品溶液色譜圖中,在與對照品色譜相應的位置上,有相同的色譜峰,而陰性溶液在待測組分處無干擾。
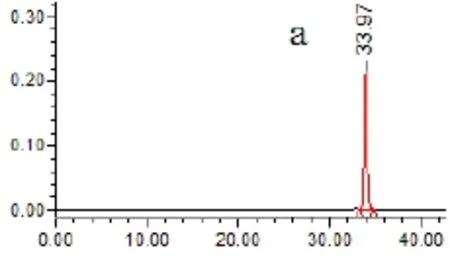
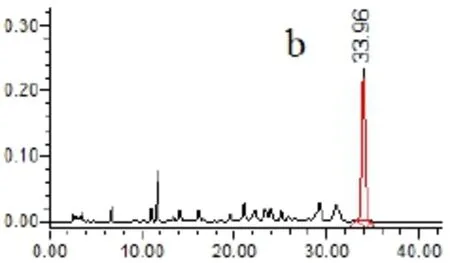
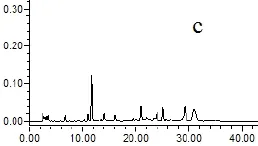
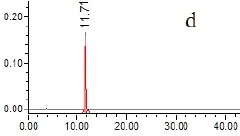
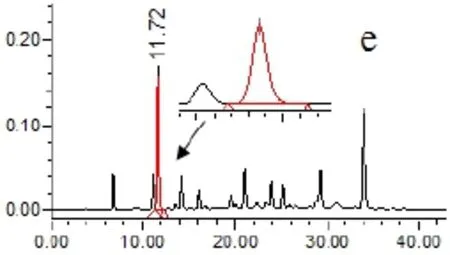

圖1 HPLC圖
2.4.6 標準曲線的繪制 取黃芩苷對照品溶液(每 1mL含 238.2μg)2.0、4.0、6.0、8.0、10.0mL,分別置10mL容量瓶中,加甲醇至刻度,配制成5種不同濃度的黃芩苷對照品溶液,取綠原酸對照品溶液2.0、4.0、6.0、8.0、10.0mL,分別置10mL容量瓶中,加甲醇至刻度,配制成5種不同濃度的綠原酸對照品溶液,取上不同濃度的黃芩苷和綠原酸對照品溶液,分別依法測定其峰面積。以對照品峰面積為縱坐標,進樣量為橫坐標,得黃芩苷回歸方程為:Y=3.10E+06X-1.56E+05(R=0.9999);綠原酸回歸方程為:Y=1.28E+06X-5.67E+04(R=0.9999),結果表明黃芩苷進樣量在0.4764~2.382 μg,綠原酸在0.4216~2.108μg成線性關系。
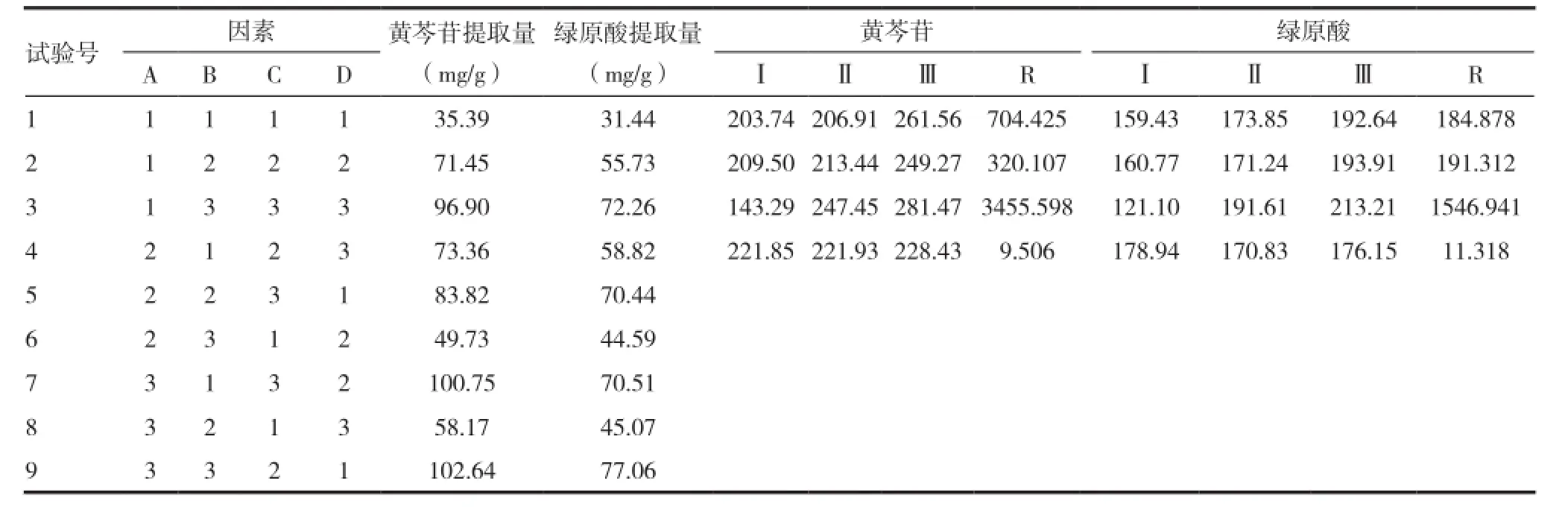
表2 正交試驗表及結果

表3 方差分析表
2.4.7 重復性試驗 重復性試驗 取黃芩苷和綠原酸對照品等體積混合溶液,連續測定6次,每次進樣5μL,測定峰面積,計算得其黃芩苷RSD為0.6%,綠原酸RSD為0.7%。結果表明,儀器精密度良好。
2.4.8 穩定性試驗 取供試品依法測定,于0、2、4、6、8、10、12h各進樣1次,測定黃芩苷、綠原酸峰面積,計算得黃芩苷RSD為1.2%,綠原酸RSD為1.1%。結果表明,供試品溶液在12 h內穩定。
2.5 工藝優選
工藝優選:以黃芩苷和綠原酸的正交實驗提取含量為指標,計算試驗結果(黃芩苷和綠原酸的提取量計算方法為:提取量=供試品溶液濃度×稀釋倍數×水煎液體積÷投料藥材重量),結果見表2~3。
結果分析:以黃芩苷為考察指標,因素A和B有統計學意義(P<0.05),C因素有統計學意義(P<0.01);以綠原酸為考察指標,因素A和B無統計學意義(P>0.05),C因素有統計學意義(P<0.01);另外,從結果中可以看出以黃芩苷和綠原酸為考察指標結果基本一致,因此,根據正交試驗直觀分析因素A、因素B和因素C均選擇水平3。最佳提取工藝為A3B3C3。即加12倍量水,煎煮3次,每次2h。綜合生產實際狀況,C因素2水平和3水平相差不大,可以考慮選擇水平2,即加12倍量水,煎煮2次,每次2h,即正交實驗9的提取方法,該方法屬于正交實驗,可以不用驗證。因此可以驗證A3B3C3的提取結果,根據結果確定最終提取工藝。
2.6 工藝驗證試驗
稱取3份處方藥材,按照加12倍量水,煎煮2次,每次2h的工藝提取,用上述方法測定黃芩苷和綠原酸的提取量。結果,樣品中黃芩苷的提取量分別 為:105.7、103.6、108.0mg/g(RSD=2.08%,n=3),樣品中綠原酸的提取量分別為:80.2、78.6、81.5mg/g(RSD=1.81%,n=3),均高于正交實驗的最高含量,但是與正交9的結果相差不大,因此最佳工藝定為:加12倍量水,煎煮2次,每次2h,并認為該工藝可行。
3 討論
本實驗采用HPLC同時檢測黃芩苷、綠原酸的含量,根據文獻[9-13]采用甲醇-(0.1%、0.2%、0.4%)磷酸溶液、甲醇-冰醋酸溶液、乙腈-0.1%磷酸溶液、乙腈-0.4%磷酸溶液為流動相,經過實驗對比,最終流動相為甲醇-0.4%磷酸梯度洗脫,目標峰達到較好的基線分離,且保留時間適宜。
根據文獻提取工藝采用水煎煮提取,以君藥黃芩和金銀花中黃芩苷、綠原酸的含量為考察指標,文獻多采用綜合加權評分法[14-15],但是本實驗兩種成分的結果相一致,因此對于正交實驗結果不再進行綜合評分法處理,而是采用分別單獨處理。
[1] 饒小勇,何雙鳳,吳志鵬,等.抗流感口服液的提取工藝優選[J].中國實驗方劑學雜志,2013,19(4):31-34.
[2] 王倩倩,王錦玉,馬振山,等.星點設計-效應面法優化解毒通絡顆粒的提取工藝[J].中國實驗方劑學雜志,2014,20(9):15-18.
[3] 鄭勇鳳,王佳婧,傅超美,等.黃芩的化學成分與藥理作用研究進展[J].中成藥,2016,38(1):141-147.
[4] 付國輝,馬香芹.黃芩的化學成分及藥理作用研究進展[J].中國當代醫藥,2015,22(22):18-20.
[5] 張小娜,童杰,周衍晶,等.忍冬屬藥材藥效成分及藥理作用研究進展[J]. 中國藥理學通報,2014,30(8):1049-1054.
[6] 龐美蓉,劉零怡,高汪磊,等.綠原酸調節糖脂代謝的作用機制研究進展[J].中草藥,2015,46(2):305-312.
[7] 韓悅.金銀花活性成分的藥理作用及其進展[J].科技經濟導刊,2016,01:148-149.
[8] 司席席,孫洪勝.復方金芩片的質量標準研究[J].中國藥房,2016,27(3):415-417.
[9] 李輝,謝新民,李騰霞,等.大自然五花飲的質量標準提高研究[J].中國藥房,2016,27(6):811-814.
[10] 景霞,徐進,孫芳,湛雯.利咽合劑的質量標準研究[J].中國藥房,2015,26(36):5141-5143.
[11] 郗洋,張熙潔,劉曉紅.HPLC法同時測定連翹敗毒丸中5種成分的含量[J].中國藥房,2016,27(6):815-818.
[12] 尹香閣,田效志,熊晶晶,等.清膽和胃丸中芍藥苷和黃芩苷含量測定的研究[J].中醫學報,2014,29(1):82-85.
[13] 于紅艷,張賓.HPLC法同時測定退黃丸中綠原酸、梔子苷的含量[J].中醫學報,2014,29(8):1170-1171.
[14] 王雪,焦連慶,于敏,等.多指標綜合加權評分法優選五味子與丹參混合醇提工藝研究[J]. 中草藥,2015,46(7):998-1001.
[15] 劉敏,楊全偉,劉新國,等.多指標加權評分正交試驗優選消痤散提取工藝[J].中西醫結合研究,2015,7(4):194-197.
Optimization of extraction technology for dureqing granule
SHI Junhan1QIN Huizhen1,2ZHENG Hong1MENG Xiangle1,2
1.First Affiliated Hospital of He'nan University of Traditional Chinese Medicine,He'nan 450000,China; 2.Pharmaceutical College of He'nan University,Kaifeng 475004,China
ObjectiveTo optimize the extraction process of dureqing granule.MethodsBaicalin and chlorogenic acid were used as indexes, HPLC was used to determined content. Agilent ZORBAX XDB-C18colume (4.6mm×150mm,5 μm), Mobile phase methanol -0.4% phosphoric acid gradient elution with flow rate of 1.0 mL/min, UV detection wavelength was 280 nm(baicalin)and 327nm(chlorogenic acid ). Amount of water added, extraction time, and extraction times were selected as the factors. L9(34)orthogonal test was designed to optimize the extraction technology of Dureqing Keli.ResultsOptimal water extraction technology was as follows : adding 12 times amount of water, reflux and extract for 2 h, three times totally.ConclusionThe optimized technology was simple and extract completely, it is applied to industrial manufacture.
Dureqing granule;Bbaicalin;Chlorogenic acid;Orthogonal design;HPLC;Extraction process
R286
B
2095-0616(2016)21-56-04
2016-08-17)
國家自然科學基金·河南省人民政府人才培養聯合基金( U1304824);中國博士后科學基金特別資助(2015T80772);中國博士后科學基金面上資助(2015M582190);河南省博士后基金項目(2014-75);濟源市科技攻關項目(15013034) ;河南省科技廳“三區”人才計劃(14104259)。
Δ2016級河南大學在讀碩士研究生
▲通訊作者
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
