基因確診肯尼迪病1例報道
2016-01-20 03:00:57230032合肥安徽省第二人民醫院神經內科李曉艷安徽醫科大學第一附屬醫院神經內科尹世杰
卒中與神經疾病 2015年6期
230032 合肥,安徽省第二人民醫院神經內科[李 震 張 樂 李 彬 王 婭 李曉艷 姜 丹];安徽醫科大學第一附屬醫院神經內科(尹世杰)
?
基因確診肯尼迪病1例報道
李震張樂李彬王婭李曉艷姜丹尹世杰
230032合肥,安徽省第二人民醫院神經內科[李震張樂李彬王婭李曉艷姜丹];安徽醫科大學第一附屬醫院神經內科(尹世杰)
【DOI】10.3969/j.issn.1007-0478.2015.06.016
Kennedy病又稱脊髓延髓肌肉萎縮癥。現將本院收治的1例經基因確診的肯尼迪病報道如下。
1病例資料
患者,男,57歲。因“漸進性四肢無力8年,加重1年余”于2015年5月5日入院。患者自8年前無明顯誘因出現雙下肢乏力及肌肉跳動感,表現為蹲起困難,上臺階困難。多次檢查心肌酶譜均顯著增高,頸椎MRI未見異常。肌電圖示神經源性損害,多家醫院考慮為“運動神經元病”。給予口服中西藥治療后患者癥狀未見好轉,下肢無力進行性加重,不能快步行走。近1年以來出現雙上肢無力,持筷夾菜困難。否認高血壓病、糖尿病史。否認家族性遺傳病史。已婚,育有二女。體健,家族中無類似患者。查體: 神清,精神可,語利。雙瞳孔等大等圓,直徑3 mm,對光反射靈敏。舌肌萎縮伴纖顫(圖1),伸舌居中。軟腭抬舉正常,懸雍垂居中,咽反射存在。肌肉無壓痛,雙側肩胛帶肌萎縮,雙側骨盆帶肌、雙手大小魚際肌萎縮及骨間肌萎縮。雙上肢近端肌力Ⅲ一級,遠端肌力Ⅲ十級,雙下肢近端肌力IV一級,遠端肌力IV十級。深淺感覺正常。四肢腱反射消失。雙側巴氏征(-)。男性乳房發育(圖2),癥狀活動和休息未見有減輕。輔助檢查:丙氨酸氨基轉移酶63 U/L、總膽固醇6.32 mmol/L、低密度脂蛋白膽固醇3.40 mmol/L、甘油三酯5.55 mmol/L、極低密度脂蛋白2.52 mmol/L。肌酶譜:天冬氨酸氨基轉移酶33 U/L、肌酸激酶879 U/L、肌酸激酶同工酶71 U/L、乳酸脫氫酶241 U/L。性激素:睪酮15.89 nmol/L。神經電生理檢查示其部分肌群時程增寬,電壓增高,重收縮時運動單位減少;雙側腋神經、股神經輕中度受損(以右側為顯)。肌肉病理檢查示所檢肌肉呈神經源性損害(圖3)。基因檢測示雄激素受體(AR)基因第一外顯子(CAG)重復序列數目為50(圖4)。
2討論
肯尼迪病于1968年由Kennedy 等首先報到。1991年Spada等發現肯尼迪病病因是Xqll-12區域的雄激素受體(AR)基因第一外顯子CAG 重復序列的數目異常增多。
肯尼迪病首發癥狀多為下肢近端肌無力和萎縮,表現為蹲起困難和上樓無力,部分有延髓下運動神經元損傷,表現為吞咽障礙、構音障礙、舌肌萎縮,少見為上肢無力為最先發病。無上運動神經元受損的表現。感覺神經功能障礙。內分泌功能障礙,表現為女性乳房化趨勢,部分有血睪酮增高。血肌酸肌酶譜增高,神經電生理檢查提示神經源性損害。感覺神經傳導波幅有顯著下降,提示感覺系統也有受累。肌活檢示其神經源性損害,偶有肌源性損害。新的歐洲神經科學聯合會(EFNS)指南將患者雄激素受體基因第1個外顯子三核苷酸CAG重復序列數目≥35作為診斷的依據。本例患者中年起病,緩慢進展,表現為肢體無力、肌萎縮、舌肌萎縮其有神經源性損害,肌活檢示其神經源性損害。且患者多次檢查血脂異常增高,通過基因測序,CAG的重復序列數目為50次。綜合以上的結果,可確診為肯尼迪病。
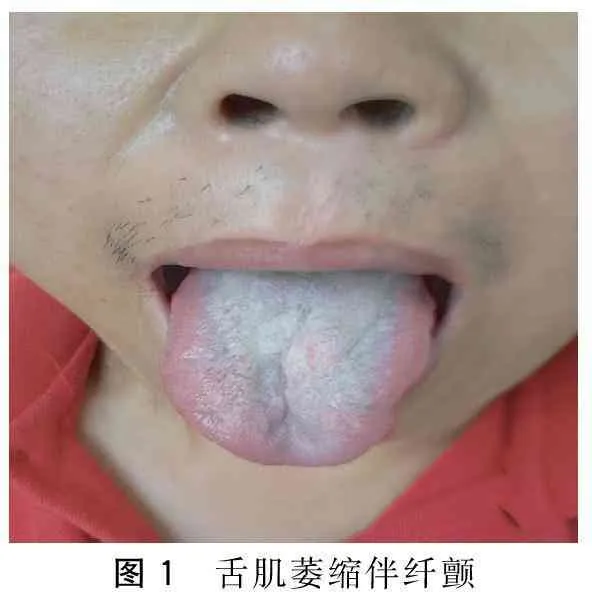
圖1 舌肌萎縮伴纖顫
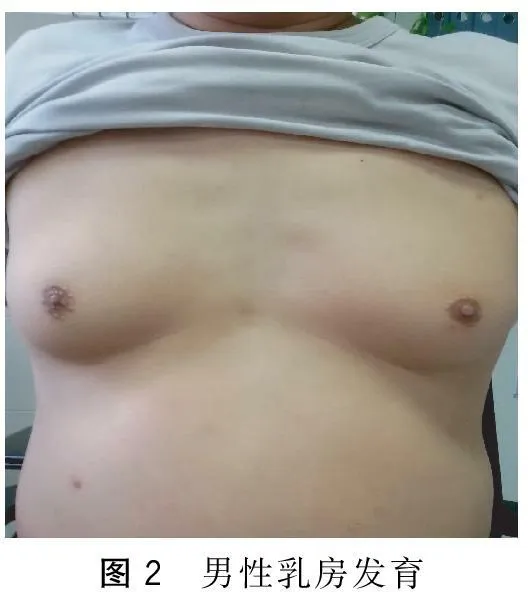
圖2 男性乳房發育
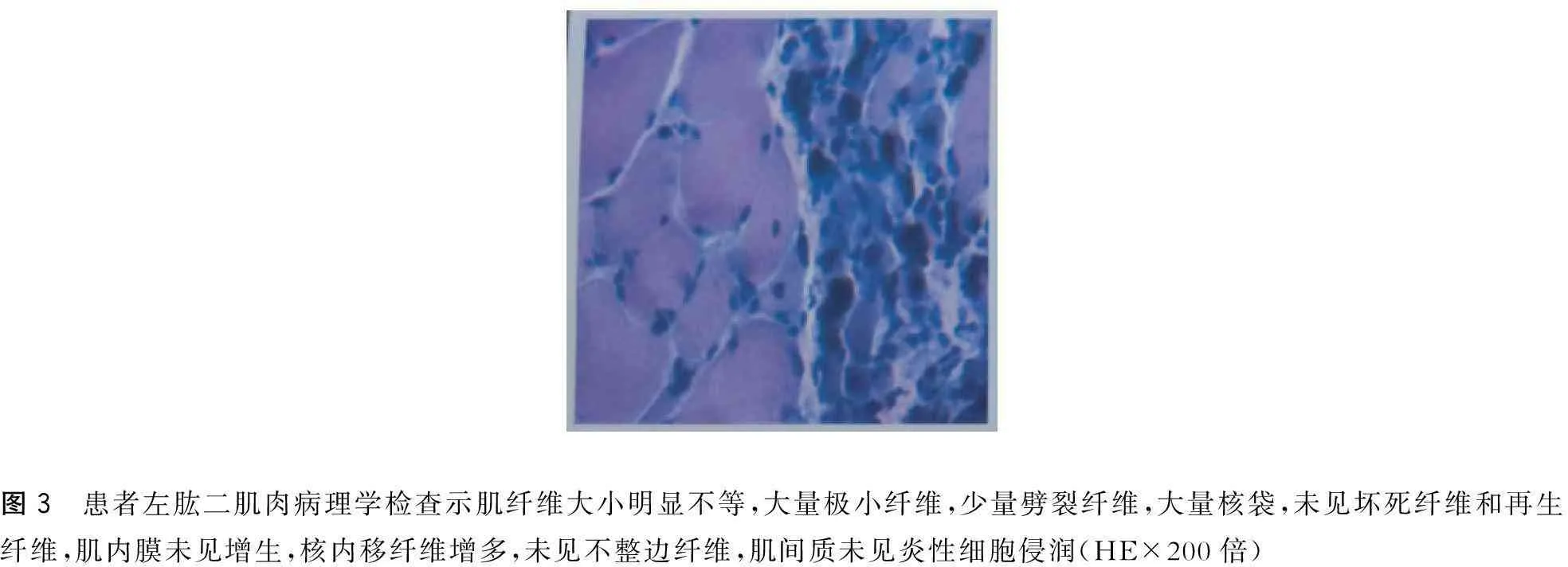
圖3 患者左肱二肌肉病理學檢查示肌纖維大小明顯不等,大量極小纖維,少量劈裂纖維,大量核袋,未見壞死纖維和再生纖維,肌內膜未見增生,核內移纖維增多,未見不整邊纖維,肌間質未見炎性細胞侵潤(HE×200倍)

圖4 患者AR基因CAG三核苷酸重復區域突變篩查示該患者AR基因的PCR產物泳動速度與正常對照者比較有明顯差異性
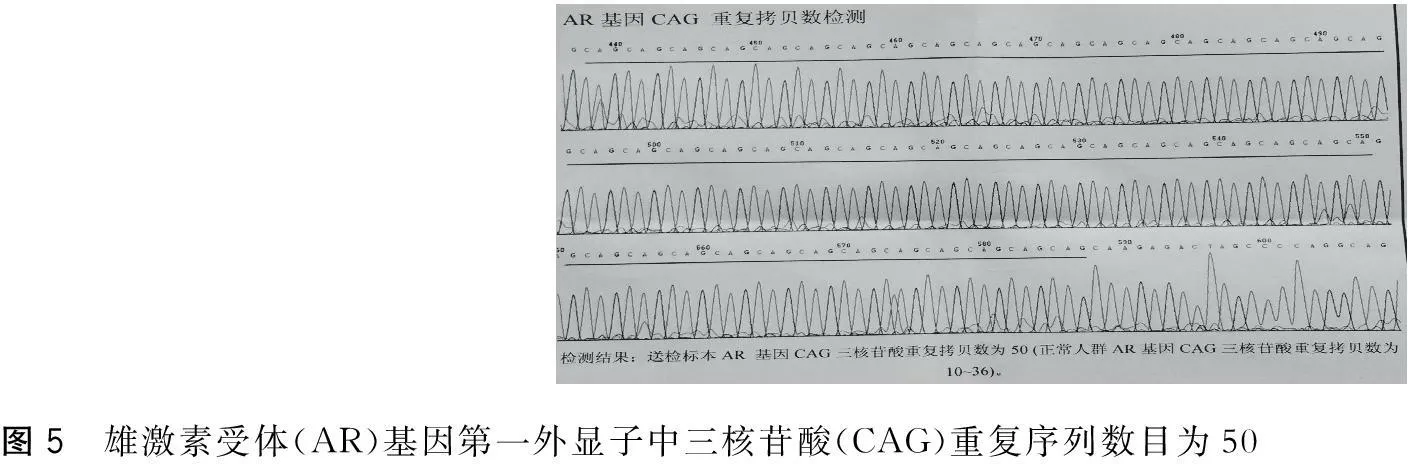
圖5 雄激素受體(AR)基因第一外顯子中三核苷酸(CAG)重復序列數目為50
KD病的發病機制不明確。有研究表明KD患者發病時間可能與CAG異常擴增次數相關,CAG異常擴增次數越多,發病時間越早。但即使重復相同CAG次數的KD患者,其發病年齡也會相差很大,提示KD發病年齡除CAG異常擴增次數之外,還存在其他的影響因素。雄激素可能是KD發病的另一個重要影響因素。
因本病臨床表現與其他神經系統變性疾病有很多相似之處,需要鑒別的有線粒體疾病、包涵體肌炎、成人起病的脊髓性肌萎縮癥、多灶性運動神經病、肌營養不良、肌萎縮側索硬化等。
目前無有效治療KD病的方法。藥物去勢治療如醋酸亮丙瑞林可延緩患者運動功能障礙和改善吞咽功能。克倫特羅可以提高患者的運動功能的情況,但有較大毒性。目前以ASC-J9為代表可望成為治療KD的新藥。
(2015-07-02收稿)
【中圖分類號】R746
【文獻標識碼】A
【文章編號】1007-0478(2015)06-0370-02
