微波輔助合成Ru配合物及其自組裝多層膜
2015-12-05 07:27:42李孔齋魏永剛
無機化學學報 2015年6期
關鍵詞:實驗
楊 麗 王 華*, 李孔齋 魏永剛 祝 星
(1昆明理工大學冶金與能源工程學院,昆明 650093) (2昆明理工大學省部共建復雜有色金屬資源清潔利用國家重點實驗室,昆明 650093)
楊麗1,2王華*,1,2李孔齋1,2魏永剛1,2祝星1,2
(1昆明理工大學冶金與能源工程學院,昆明650093) (2昆明理工大學省部共建復雜有色金屬資源清潔利用國家重點實驗室,昆明650093)
運用微波輔助合成技術制備得到對稱性釕配合物,對該配合物進行了1H NMR,ESI-MS和TG分析。該釕配合物兩端對稱的磷酸基團可通過共價鍵作用組裝到納米銦錫金屬氧化物導電玻璃(Indium Tin Oxides,ITO)表面,使ITO表面呈現親水性。利用鋯離子作為橋梁成功組裝了釕多層膜,并對該多層膜進行了循環伏安法(Cyclic Voltammetry,CV)及紫外-可見吸收光譜法(Ultraviolet-visible absorption spectrometry,UV-Vis)等光電化學分析,實驗結果表明層層自組裝過程中膜沉積均勻,在0.53 V出現可逆的氧化還原峰,在300~600 nm的紫外可見區域出現強且寬的吸收峰,表明該釕配合物具有優良的光電性能。
釕配合物;自組裝;微波輔助;光電性質
0 引 言
當今社會能源問題日益突出,尋找清潔且可持續能源是人類所面臨的關鍵性問題之一,而太陽能因其普遍、無害、巨大和長久性而受到研究者的青睞。由Gr?zel等[1-2]于1985年發明的染料敏化太陽電池(Dye-Sensitized Solar Cells,DSSCs)因其制作簡單、成本低廉、性能穩定以及對環境無污染等優點,在當今能源匱乏和環境污染嚴重等突出問題下,其研究廣受關注,已經成為太陽能電池研究的重要課題[3]。敏化劑是染料敏化太陽電池結構中的主要部分,直接影響著電池對可見光的吸收以及光生電子的產生和注入[4]。提高敏化劑的光電性能,對于太陽能電池的應用具有重要的現實及推廣意義。釕配合物由于其獨特的化學穩定性、氧化還原性、良好的激發態反應活性、和較長的激發態壽命,現已為太陽電池中首選的高效敏化劑[5-8]。
釕配合物自組裝法是根據分子的自組裝作用,在電極表面形成高度有序的分子層[9-10]。自組裝單分子膜可通過含有自由運動的端基,例如硫醇、氨基等的有機分子對電極表面改性并賦予電極表面新的功能[11-18]。釕配合物的組裝方式主要有LB膜法[19-20]、電化學法[21-23]、共價鍵合法[24]、吸附固定[25]等方法。自組裝多層膜無需復雜的儀器和設備,操作簡單,可通過分子組裝過程將不同種類和功能的構筑基元按照一定需求進行組裝,具有成膜物質豐富,成膜不受基底限制,可在分子水平控制組裝體系的結構及性質,制備的薄膜具有良好的穩定性,薄膜的組成和厚度可控等諸多優點[26-27]。
本文以RuCl3·3H2O為原料,在微波輔助條件下合成穩定性高、光電化學性能優異的對稱性苯并咪唑類釕配合物[Ru(HL)2](PF6)2記為Ru-1(如圖1所示),該釕配合物以兩端對稱的磷酸基團作為固定配體,以Zr4+為橋梁實現了釕配合物多層膜的層層自組裝。通過1H NMR、MS表征了該配合物的結構,并對該配合物形成的多層膜進行了電化學及紫外可見吸收光譜等光電化學分析。
1 實驗部分
1.1實驗試劑及儀器
測試所用的乙腈使用前加五氧化二磷 (P2O5)重蒸2次,支持電解質四丁基六氟磷酸銨(TBAPF6)使用前用乙醇(EtOH)重結晶、真空干燥,分析用溶劑為色譜純,實驗用水為超純水或二次蒸餾水,其他試劑均為市售,沒有經過進一步純化。
以核磁共振儀(JEOL-ECA500,Japan)和質譜儀(AXIMA CFR Plus MALDI-TOF,UK)測定配合物的純度和分子結構;熱分析儀 (NETZSCH STA449C,Germany)測定配合物的熱穩定性;分光光度計(U-4000,HITACHI,Japan)測定紫外可見光譜;電化學分析儀(ALS/CHI 660A,BAS,USA)測定電化學數據,測定過程中以銀硝酸銀作為參照電極,Pt作為對比電極,測定的數據進行電位補正;接觸角測量儀(CA-X,Japan)測定電極表面接觸角。
1.2釕配合物的微波輔助合成
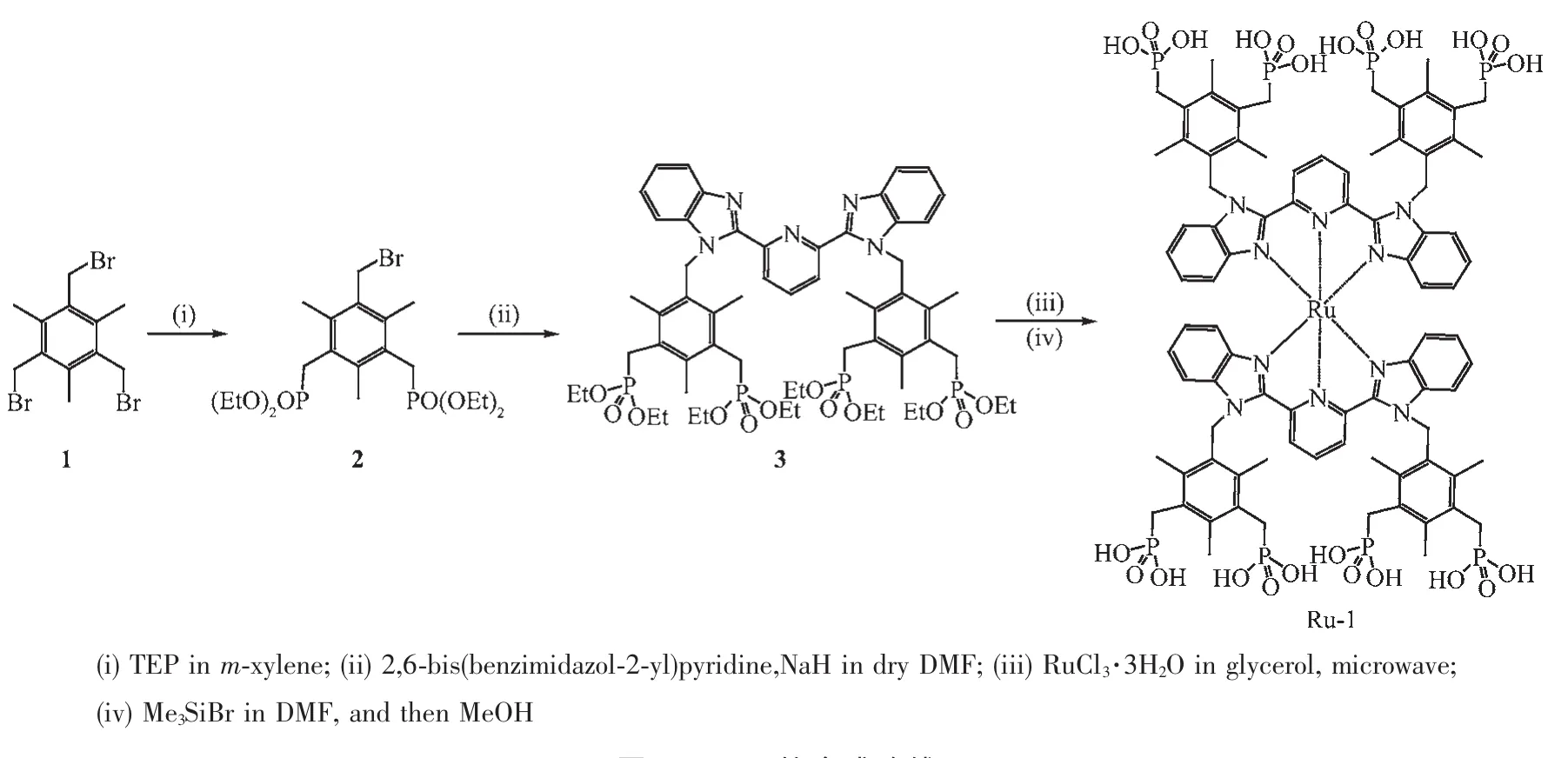
圖1 Ru-1的合成路線Fig.1 Synthetic route of Ru-1 complex
2,6-二(N-(2,4,6-甲基-3,5-二乙基膦酸酯甲基-芐基)-2-苯并咪唑基)吡啶[29]與RuCl3·3H2O在丙三醇溶液中借助微波輔助合成中間體[Ru(EtL)2] (PF6)2,最后與三甲基溴硅烷發生去保護反應得到目標配產物Ru-1,合成路線如圖1所示。
1.2.11-溴甲基-3,5-二(二乙基膦酸酯甲基)-2,4,6-三甲基苯(化合物2)
在反應器中將5 g(12.53 mmol)2,4,6-三溴甲基三甲基苯(化合物1)溶解到25 mL間二甲苯中,將4.57 g(25.06 mmol)磷酸三乙酯溶解到25 mL間二甲苯中后滴入反應器中,在90℃下加熱攪拌20 h后減壓蒸餾除去溶劑得到粗產物,所得粗產物的提純采用柱色譜法,固定相使用63~210 μm的球形硅膠,固定相直徑為7.5 cm,高度為5 cm,流動相為EtOAc溶液,經柱色譜法進行提純得到淺黃色產物2。1H NMR(CDCl3,500 MHz)δ:4.69(2H,s),3.96(8H,m),3.38(4H,d,J=12.7Hz),2.63(3H,s),2.49(6H,s),1.22(12H,t,J=7.5 Hz)。MS(MALDI-TOF,CH2Cl2):m/z=513.15,[M]計算值為 513.346 2,其中 M= C20H35BrO6P2。元素分析,計算值(%):C 46.79,H 6.89;實驗值(%):C 46.65,H 6.97。
1.2.22,6-二(N-(2,4,6-甲基-3,5-二乙基膦酸酯甲基-芐基)-2-苯并咪唑基)吡啶(配體3,簡記為EtL)
在氮氣氣氛下,將經正戊烷洗滌后的1.08 g (27.12 mmol)30%NaH懸浮于20 mL無水 DMF中,加入1.78 g(5.71 mmol)2,6-二[2-苯并咪唑基]吡啶,在70℃下加熱攪拌10 h,在此期間懸浮物漸漸溶解得到澄清的黃色溶液,將反應液轉移到恒壓滴液漏斗中,在室溫下逐滴滴加到5.84 g(11.42 mmol)化合物2中,在70℃下加熱攪拌20 h,冷卻至室溫后加入3 mL甲醇,減壓蒸餾除去溶劑得到白色粗產物。所得的粗產物采用柱色譜法提純,固定相使用63~210 μm的球形硅膠,固定相直徑為7.5 cm,高度為6 cm,流動相為丙酮溶液,經柱色譜法進行提純得到黃色產物3。1H NMR(CDCl3,500 MHz)δ:8.47(2H,d,J=8.5Hz),8.19(1H,t,J=7.2Hz),7.79(2H,d,J=7.3 Hz),7.13(2H,t,J=7.9 Hz),6.96(2H,t,J=7.7 Hz),6.78(2H,d,J=8.9Hz),6.12(4H,s),3.95~3.86(16H,m),3.27(8H,d,J=22.2 Hz),2.74(6H,s),2.20(12H,s),1.12(24H,t,J=7.64 Hz)。MS(MALDITOF,CH2Cl2):m/z=1176.19,[M]計算值為1176.4922,其中M=C59H81N5O12P4。元素分析,計算值(%):C 60.23, H 6.95,N 5.95;實驗值(%):C 60.09,H 6.78,N 6.13。
1.2.3中間配合物[Ru(EtL)2](PF6)2
實驗前,取0.26 g(1.25 mmol)RuCl3·3H2O進行真空干燥0.5 h,實驗時在氮氣氣氛下,將已干燥的RuCl3·3H2O溶解于25 mL的丙三醇溶液中,加入2.94 g,2.5 mmol的EtL,微波加熱至250℃后恒溫反應3 min,得到的溶液冷卻至室溫,加入過量的KPF6溶液后傾入水中,得到的固體經過濾、干燥后得到紫色粗產物。所得粗產物的提純采用葡聚糖凝膠凝膠色譜法,固定相使用Sephadex LH-20,固定相直徑為3 cm高度為15 cm,流動相為50/50(V/V)的MeOH/MeCN溶液,經凝膠色譜法提純后得到紫色配合物中間配合物[Ru(EtL)2](PF6)2。1H NMR (CDCl3,500 MHz)δ:8.79(4H,d,J=8.2 Hz),8.47(2H,t,J=7.7 Hz),7.92(4H,d,J=7.1 Hz),7.22(4H,t,J=7.6 Hz),6.59(4H,m),6.41(4H,d,J=8.1 Hz),6.05(8H,s),4.36~3.98(32H,m),3.53(16H,d,J=22.2 Hz),2.85 (12H,s),2.33(24H,s),1.28(48H,m)。MS(ESI-TOF,CH3CN):m/z=1 226.42,[M]計算值為1 226.73,其中M=C118H162N10O24P8Ru。元素分析, 計算值(%):C 57.61,H 5.95,N 5.11;實驗值(%):C 51.31,H 6.05,N 5.28。
1.2.4目標釕配合物[Ru(HL)2](PF6)2(簡記為Ru-1)
氮氣氣氛下,將1.96 g,0.80 mmol的[Ru(EtL)2] (PF6)2溶解到80 mL的無水DMF中,分3次滴加27.61 mL,208 mmol的Me3SiBr,每次滴加間隔30 min,在室溫下攪拌45 h后加入15 mL的MeOH,在室溫下攪拌10 h后減壓除去MeOH和DMF,經MeCN洗滌后溶于氨水中,加入過量KPF6溶液,用鹽酸調節pH值直至產生紫色 沉淀,過濾即得紫色產物[Ru(HL)2](PF6)2。1H NMR(DMSO-d6,500 MHz)δ:8.73(4H,d,J=8.9 Hz),8.42(2H,t,J=7.9 Hz),7.96(4H,d,J=7.3 Hz),7.27(4H,t,J=7.0 Hz),6.52(4H,m),6.44 (4H,d,J=8.8 Hz),6.13(8H,s),3.57(16H,d,J=20.3 Hz),2.80(12H,s),2.27(24H,s)。元素分析,計算值(%):C 45.02,H 4.30,N 6.10;實驗值 (%):44.91,H 4.21,N 6.22。
1.3電極制備
釕配合物溶液的配制:在干凈的燒杯中加入10 mL超純水,用氨水調節pH值至10,稱取2.29 mg釕配合物Ru-1溶解于溶液中,用HCl調節pH值至5后,加入超純水至20 mL,即得50 μmol·L-1的釕配合物溶液。
ITO導電玻璃的前處理:ITO的表面處理采用Radio Corporation of America(RCA)方法[28]。將NH3、H2O2和超純水按1∶1∶5的體積比混合配制RCA溶液,將ITO基片浸沒于RCA溶液中正面朝外,輕微震蕩除去氣泡后移至已盛有熱水的水浴鍋內,在80℃加熱1 h后取出ITO基片用超純水洗凈,氮氣吹干。
電極制備:將經表面處理后的ITO基片浸沒于釕配合物溶液中正面朝外,輕微震蕩除去氣泡,在室溫下浸漬3 h后取出ITO基片用超純水洗凈、氮氣吹干,該電極為陽極。
以伏安法為中心的電化學測定方法采用三電極系統,經Ru-1修飾的ITO電極作為工作電極,Ag/AgNO3為參比電極,Pt線作為對比電極,使用前用砂紙將表面磨光,用重鉻酸混合液、熱硝酸等清洗,最后用水沖洗干凈。支持電解質為TBAPF6,溶劑為無水無氧乙腈,濃度為1 mol·L-1,測試的最后加入二茂鐵[Fe(C5H5)2],進行電位校正。
2 結果與討論
2.1釕配合物的熱穩定性分析
當釕配合物作為染料敏化劑使用時,其穩定性直接決定著染料敏化太陽能電池的穩定性,提高染料的穩定性和提高光電轉換效率同樣重要,因此有必要對釕配合物進行熱穩定性分析。采用熱分析儀對Ru-1的熱穩定性進行測定,每次實驗取9 mg的樣品置于氧化鋁坩堝中,在空氣流中進行實驗,升溫速率為10℃·min-1,溫度范圍為40~800℃,系統自動采集數據,得到樣品的TG數據和DSC數據,根據實驗數據繪制出釕配合物Ru-1的TG-DSC曲線,如圖2所示。
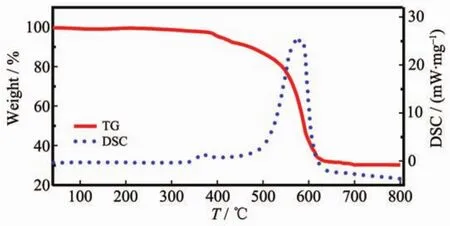
圖2 Ru-1的TG-DSC曲線Fig.2 TG-DSC curves of Ru-1 complex
從圖2中可以看出,Ru-1在400℃前很穩定,在400~530℃溫度區間內開始緩慢分解,這是由于Ru-1分子上的羧基氧化成CO2。530~600℃是Ru-1主要分解區間,失重率約占整個溫度區間的60%,這主要是由于伴隨溫度升高,配體發生劇烈氧化,Ru-1不斷分解。600℃以后重量仍在32%以上,說明樣品中仍有難熱解的基團存在。配體的DSC曲線中,380℃附近較小的吸熱峰說明有輕微的脫羧反應,而Ru-1分解反應所形成的明顯吸熱峰約在580℃出現。通過對Ru-1的熱分析過程可以看出,Ru-1在一個較寬的溫度范圍內是非常穩定的,其熱穩定性完全能夠滿足染料敏化太陽電池對敏化劑熱穩定性的要求。
2.2接觸角測定
Ru-1修飾的ITO電極表面接觸角如圖3所示,未經處理的ITO基片表面接觸角為86.3°,說明潔凈ITO表面的親水性較低;經表面RCA處理后的ITO基片表面的接觸角為66.4°,明顯提高了ITO表面的親水性;自組裝釕配合物單分子膜后的ITO基片的表面接觸角為51.8°,表面親水性升高,這是因為Ru-1分子中兩端對稱的親水基,一端固定到ITO界面,將另一端親水基暴露在表面,使組裝Ru-1單分子膜后的ITO基片表面呈現出親水性,同時,接觸角的改變也證明了ITO上已成功組裝上Ru-1單分子膜。

圖3 ITO基片的表面接觸角(a)未經處理,(b)RCA表面處理后,(c)Ru-1修飾后Fig.3 Contact angle of ITO surface(a)bare ITO,(b)after RCA treatment,(c)Ru-1 modified
2.3釕配合物多層膜的層層自組裝
制備的Ru-1分子兩端為對稱的膦酸基團,其中一組膦酸基團通過共價鍵自組裝到ITO表面,另一組磷酸基裸露在外層,通過Zr4+-磷酸酯鍵可形成具有氧化還原性能的釕配合物多層膜,自組裝單層膜及多層膜的結構如圖4所示。
通過形成Zr4+-磷酸酯層來實現釕配合物Ru-1層層自組裝多層膜的組裝示意圖如圖5所示。首先將ITO基片浸沒于RCA溶液中正面朝外,輕微震蕩除去氣泡后移至已盛有熱水的水浴鍋內,在80℃加熱1 h后取出ITO基片用超純水洗凈,氮氣吹干。然后將經表面處理后的ITO基片浸沒于濃度為50 μmol·L-1的Ru-1溶液中正面朝外,輕微震蕩除去氣泡,在室溫下浸漬3 h后取出ITO基片用超純水洗凈氮氣吹干,得到單層膜修飾的ITO基片;將單層膜修飾的ITO基片浸沒于濃度為20 mmol·L-1的ZrOCl2溶液中輕微震蕩除去氣泡,在室溫下浸漬30 min,使Zr4+連接到膦酸基團上,取出ITO基片,用超純水洗凈氮氣吹干后再次浸沒于濃度為50 μmol·L-1的Ru-1溶液中,使Ru-1中的膦酸基團連接到Zr4+上,重復以上步驟得到層數可控的釕配合物多層膜。
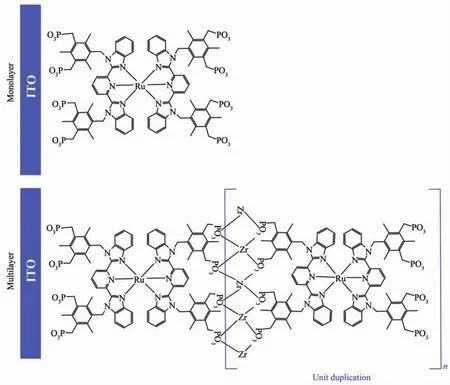
圖4 Ru-1在ITO表面的自組裝單層膜結構及通過Zr-磷酸酯鍵形成的多層膜結構Fig.4 Proposed surface-tethered structures for the Ru-1 monolayer on ITO;And multilayer of Ru-1 connected by Zr-phosphonate bond on ITO
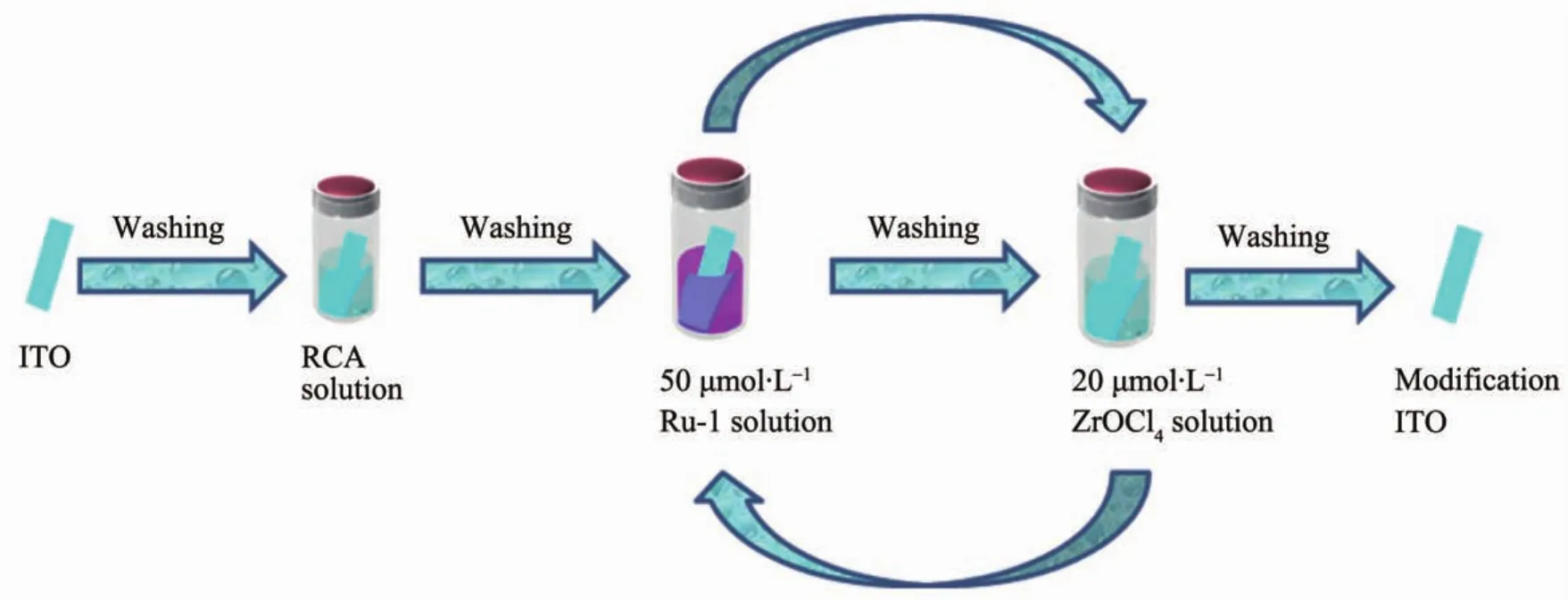
圖5 Ru-1多層膜的組裝示意圖Fig.5 Schematic of the multilayered assembly film of Ru-1 complexs fabrication process
2.4電化學研究
Ru-1單層膜在ITO基板上的吸附關系通過循環伏安法判定,參數設置如下:初始電位為0 V;高電位為1.2 V;低電位為0 V;掃描次數為6次;等待時間為3~5 s;靈敏度選擇為10 μA;濾波參數為50 Hz;放大倍數1;掃描速度(單位為V·s-1)根據實驗需要分別設定為:0.1,0.2,0.3,0.4,0.5。測定過程中以0.1 mol·L-1的TBAPF6溶液(溶劑為無水乙腈)作為電解質,使用前在真空下干燥3 h。實驗以僅覆蓋一層Ru-1的ITO(ITO/Ru-1)為工作電極,Ag/AgNO3作為參照電極,Pt線作為對比電極,進行CV測定前先通入20 min氮氣去除溶液中的氧,測定時液面保持氮氣氣氛,測定的數據進行電位補正,實驗得到的Ru-1單層膜修飾的ITO電極的伏安圖如圖6所示。
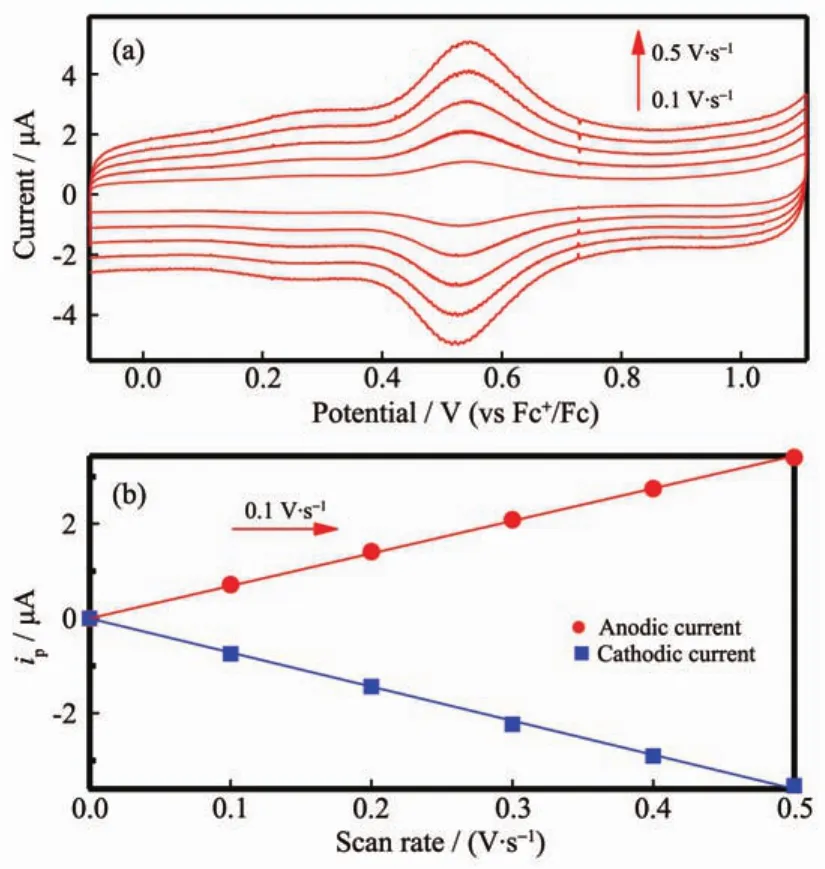
圖6 (a)Ru-1單層膜修飾的ITO電極的循環伏安曲線;(b)掃描速度與電流關系圖Fig.6 (a)Typical cyclic voltammogram of Ru-1 on an ITO;(b)Dependence of the peak current on the scan rate
循環伏安實驗表明,釕配合物Ru-1修飾的ITO電極表現出可逆的氧化還原過程,在0.53 V(vs Fc+/ Fc參比電極)出現一對氧化還原峰,可歸屬為Ru(Ⅱ/Ⅲ)的氧化還原過程,該電位高于I-/I3-氧化還原電對的電位[30],由此可說明當Ru-1修飾的ITO電極用于染料敏化太陽能電池時,將為電子的傳遞和染料的高效再生提供驅動力。從圖6(a)中可以看出不同的掃描速度下具有相似的循環曲線,且電流值隨掃描速度的增加而增加,從圖6(b)中可以清晰地看到電流與掃描速度成直線增長關系,陽極電流與陰極電流均滿足ip∝V關系,證明Ru-1單層膜與ITO基板的關系為吸附關系,即Ru-1分子已成功自組裝到ITO電極表面。氧化還原過程中的電荷轉移與釕單層膜密度成正比,公式(1)為電極表面的覆蓋量的計算公式,通過公式(1)計算得到Ru-1單層膜修飾的ITO電極的最大表面覆蓋量為2.76×10-11mol· cm-2。

其中Γ(mol·cm-2)、Q、F、n和A分別為表面覆蓋量、電荷量、Faraday常數(96 485 C·mol-1)、電荷轉移數和工作電極的電活性面積(0.26 cm2)。
為研究Ru-1單層膜的穩定性,將經Ru-1修飾的ITO浸漬于DMF溶液中,隨著解析時間的增加,Ru-1分子逐漸脫離ITO表面擴散到溶液體系中,表現為在不同解析時間下Ru-1單層膜的電流呈現出緩慢下降的趨勢。浸漬一段時間后取出電極,用新鮮DMF徹底清洗除去電極表面解吸出的Ru-1分子,氮氣吹干后重新組裝三電極系統,測定不同浸漬時間下的循環伏安曲線,掃描速度設定為0.1 V· s-1,測定的數據進行電位補正。得到Ru-1解吸率與解吸時間的關系圖,如圖7所示。
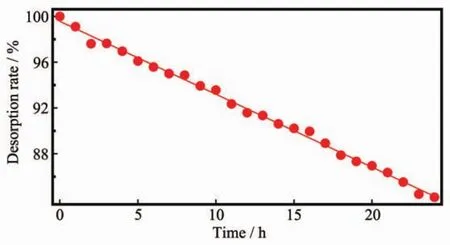
圖7 Ru-1在ITO電極上的解吸率與解吸時間的關系圖Fig.7 Dependence of desorption rate on time for Ru-1 complex on ITO surface
從圖7中可以清晰地看出Ru-1分子膜表現出優秀的穩定性,當解吸時間為24 h時,Ru-1單層膜的覆蓋率依舊高于83%,也就是說將Ru-1單層膜修飾的ITO電極浸漬于DMF溶液中長達1 d也不會導致Ru-1從ITO電極表面脫落,其解吸常數k= 1.91×10-6s-1,與已有文獻對比,Ru-1分子的解吸速度較慢,說明Ru-1單層膜在一個較寬的時間范圍內是穩定的。需要說明的是經Ru-1單層膜修飾的ITO電極浸漬于溶解性較差的溶劑或水溶液中時,其解吸速率將會更低。
多層釕配合物 (Ru-1)n的層層自組裝可通過循環伏安法進行監測,其結果如圖8(a)所示。多層膜修飾的ITO電極表現出可逆的氧化還原過程,不同層數修飾的ITO電極具有相似的循環伏安曲線,在0.53 V(vs Fc+/Fc參比電極)出現Ru(Ⅱ/Ⅲ)的氧化還原峰,且電流值隨多層膜層數的增加而增加,每一層的電流與掃描速度均成直線增長關系,陽極電流與陰極電流均滿足ip∝V關系,證明多層膜(Ru-1)n (n=0~5)與ITO基板的關系均為吸附關系,說明多層膜已組裝到ITO電極上。通過公式(1)計算出每一層膜的表面覆蓋量,并繪制出ITO電極表面的多層膜層數與表面覆蓋量的關系圖如圖8(b)所示。從圖8 (b)中可以清晰地看到表面覆蓋量隨多層膜層數的增加呈現出直線增長的關系,說明在層層自組裝過程中每一層釕配合物膜都是均勻生長的。
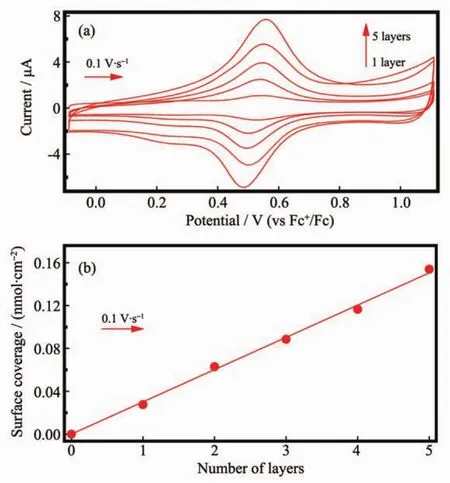
圖8 (a)多層Ru-1修飾的ITO電極的循環伏安曲線;(b)ITO電極表面的多層膜層數與表面覆蓋量的關系圖Fig.8 (a)Cyclic voltammograms of successive LbL growth of (Ru-1)n layers on ITO and(b)surface coverage versus number of layers for(Ru-1)n alternative-layer film on ITO and“n”stands for the number of the layers(n=0~5)
2.5紫外可見吸收光譜分析
利用紫外-可見光吸收儀可以很好地研究薄膜對光的吸收性能。Ru-1自組裝多層膜(Ru-1)n的紫外-可見吸收光譜如圖9所示。
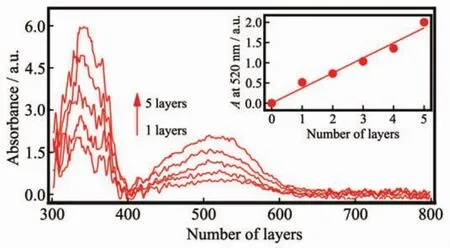
圖9 自組裝多層膜(Ru-1)n的紫外可見吸收光譜Fig.9 UV-Vis spectra monitoring of successive LBL fabrication of(Ru-1)n
從圖9中可以看出自組裝多層膜(Ru-1)n的UV曲線呈現出典型的釕配合物特征吸收峰,300~600 nm區間出現的兩個吸收譜帶證明(Ru-1)n多層膜已自組裝到ITO電極上。330 nm附近對應的強吸收帶是基于配體自旋允許的π-π*電子躍遷產生的吸收。520 nm附近的吸收帶則歸屬于配合物中金屬-配體間的電荷轉移激發態,即MLCT(metal-to-ligand charge transfer transition)躍遷吸收,其中MLCT呈現出較寬的峰,這可能是由染料分子結構決定的,Ru-1在較寬的紫外可見區域表現出較強的吸收,這對太陽光的捕獲效率是十分有利的[31]。從圖9中的插圖可以清晰地看到隨著層數的增加,MLCT躍遷吸收譜帶呈現出直線增長的趨勢,說明在層層自組裝過程中,隨著自組裝層數的增加每一層的釕配合物膜都是均勻生長的,這與電化學分析的結果是一致的。
3 結 論
[1]Desilvestro J,Gr?tzel M,Kavan L,et al.J.Am.Chem.Soc.,1985,107:2988-2990
[2]O′Regan B,Grtzel M.Nature,1991,352:737-740
[3]Vougioukalakis G,Philippopoulos A,Stergiopoulos T.Coord. Chem.Rev.,2011,255:2602-2621
[4]Wang P,Zakeeruddin S M,Moser J E,et al.Nat.Mater., 2003,2:402-407
[5]Gao S Y,Li X,Yang C P,et al.J.Solid State Chem.,2006, 179:1407-1414
[6]JIANG Cai-Wu(蔣才武),CHAO Hui(巢暉),LI Run-Hua(李潤華),et al.Acta Chim.Sinica(化學學報),2002,60:65-70
[7]Sussuehi E M,Lima A A,Giovani W F.Polyhedron,2006, 25:1457-1463
[8]GUO Chui-Lian(郭翠蓮),WU Yue(吳悅),ZHOU Yi-Ming(周益明),et al.Chinese J.Inorg.Chem.(無機化學學報),2007, 10:1771-1776
[9]SHAO Hui-Bo(邵會波),YU Hua-Zhong(于化中),ZHANG Hao-Li(張浩力),et al.Acta Phys.-Chim.Sin.(物理化學學報),1998,14:772-777
[10]Mezzenga R,Ruokolainen J,Fredrickson G H,et al.Scince, 2003,299:1872-874
[11]Parak W J.Science,2011,334:1359-1360
[12]ZHANG Yu-Qi(張玉琦),GAO Li-Hua(高麗華),DUAN Zhi-Ming(段智明),et al.Acta Chim.Sinica(化學學報),2004, 62:738-741
[13]Li S,Li J,Chen S,et al.Mater.Chem.Phys.,2013,142:513 -520
[14]WANG Cheng(王程),SHI Hui-Dheng(施惠生),LI Yan(李艷),et al.Chinese J.Inorg.Chem.(無機化學學報),2011, 27:2239-2244
[15]Dubey A,Mishra A,Min J,et al.Inorg.Chim.Acta,2014, 423:326-331
[16]Schmitt J,Raatz A,Dietrich F,et al.CIRP Ann-Manuf. Technol.,2014,63:9-12
[17]Yang S,Jin X,Liu K S.Particuology,2013,11:361-370
[18]Park S,Lim J H,Chung S W,et al.Science,2004,303:348-351
[19]Du Y,Wei H,Kang J Z,et al.Anal.Chem.,2005,77:7993-7993
[20]Fukuda N,Mitsuishi M,Aoki A,et al.J.Phys.Chem.B, 2002,106:7048-7052
[21]Decher G.Science,1997,277:1232-1237
[22]Geneste F,Moinet C.J.Electroanal.Chem.,2006,594:105-110
[23]He P,Bayachou M.Langmuir,2005,21:6086-6092
[24]Pinheiro S O,Deousa J R,Santiago M O,et al.Inorg.Chim. Acta,2006,359:391-401
[25]Lahav M,Heleg Shabtai V,Wasserman J,et al.J.Am. Chem.Soc.,2000,122:11480-11487
[26]Zhou Y,Yan D.Chem.Commun.,2009,10:1172-1188
[27]Hong H G,Mallouk T E.Langmuir,1991,7:362-2369
[28]Donley C,Dunphy D,Paine D,et al.Langmuir,2001,18: 450-457
[29]Kobayashi K,Tonegawa N,Fujii S,et al.Langmuir,2008, 24:13203-13211
[30]Erten-Ela S,Sogut S,Ocakoglu K.Mater.Sci.Semicond. Process.,2014,23:159-166
[31]Kalyanasundaram K,Nazeeruddin M K,Gritzel M.Inorg. Chim.Acta,1992,198:831-839
Microwave-Assisted Synthesis of RuComplex and Self-Assembled Multilayer Film Formed by Alternating Layers of the Ru Complex
YANG Li1,2WANG Hua*,1,2LI Kong-Zhai1,2WEI Yong-Gang1,2ZHU Xing1,2
(1Faculty of Metallurgical and Energy Engineering,Kunming University of Science and Technology,Kunming 650093,China) (2State Key Laboratory of Complex Nonferrous Metal Resources Clean Utilization,Kunming University of Science and Technology,Kunming 650093,China)
A symmetrical ruthenium complex bearing phosphonic acid was prepared under the microwave irradiation and fully characterized by1H NMR,ESI-MS and thermo gravimetric(TG)analysis.For the Ru complex,the phosphonic groups were selectively attached to the ITO through covalent interaction,resulting in the hydrophilic surface due to the appearance of the exposed other phosphonic-acid groups on the top.The Layer by layer(LBL)growth of molecular units was used to fabricate redox-active films of Ru complex,which was followed by the formation of Zr4+-phosphonate layer.The buildup of the films was followed by monitoring cyclic voltammetry (CV)and ultraviolet-visible absorption spectrometry(UV-Vis)measurements.The results showed the electrode modified by multilayered film displayed reversible redox processes,and the Ru(Ⅱ/Ⅲ)oxidative peak was observed at+0.53 V.The plots of surface coverage versus number of layers show a linear relationship,which means a uniform layer structure is formed during the LBL process.UV-Vis spectra shows the prepared Ru film have strong absorptions between 300 nm and 600 nm.These findings show the Ru complex has good photoelectric properties.
ruthenium complex;self-assembled;microwave-assisted;photoelectric character
O614.82+1
A
1001-4861(2015)06-1131-08
10.11862/CJIC.2015.151
2014-12-12。收修改稿日期:2015-02-07。
國家自然科學基金(No.51174105)和國家自然基金面上項目(No.51374004)資助。
*通訊聯系人。E-mail:wanghuaheat@hotmail.com
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55