等離子光譜法測定巖石中微量元素的相關研究
2015-12-03 09:04:16盛曉波
現代鹽化工 2015年6期
關鍵詞:實驗
盛曉波
(華東冶金地質勘查局中心實驗室,安徽 合肥 230088)
一、等離子光譜法概述
電感耦合等離子體離子發射光譜(ICP-MS)法是一種檢測線光譜的方法,其中MS是由激發態的電子通過去激發過程產生的線光譜組成的。電子得到激發后,便成為了活躍性的激發態,其去激發的過程,也就是由激發態向低激發態或者基態躍遷的過程中會發射出譜線。對于每一個元素而言,線光譜都具有唯一性。因此,分析者利用色散系統對光譜進行色散處理,然后選定一個具體的分析線,就可以對這一色譜進行定性和定量的分析。
二、實驗部分
1、儀器和試劑
儀器:Prodigy型全譜直讀電感耦合等離子體原子發射光譜儀;CID檢測器。
工作條件:功率1.1 kW,冷卻氣(Ar)流量20 L/rain,輔助氣流量0.2 L/min,霧化氣壓力0.2 MPa,溶液提升量1.2 mL/min;觀測方式為水平觀測,積分時間20 s。
測定工作使用的電子天平CP225D:量程0 g~220 g,最小分度值0.00001 g。
測量使用的試劑:硝酸、高氯酸、氫氟酸和鹽酸均為優級純;實驗用水為經Milli-Q純化水處理系統處理過的超純水,氬氣純度大于99.9%。
實驗采用的是國家一級的標準物質:銅礦石、鉛礦石、鋅礦石、花崗巖、多金屬礦石等。
等離子體光譜測定的各校準元素標準溶液的濃度及其組合見表1。
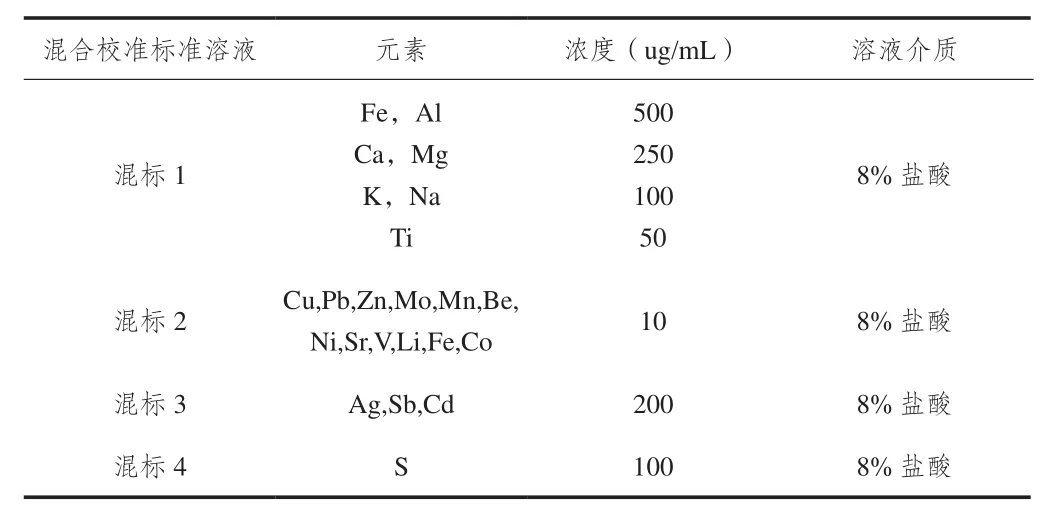
表1. 各元素校準標準溶液濃度及組合方式
2、樣品分解
選取粒子直徑小于74um的巖石樣本,置于105℃烘箱中烘烤3h,取出后置于干燥容器內冷卻至室溫,準確稱取0.12500 g巖石樣品,放于50mL的聚四氟乙烯燒杯內,向燒杯中加入5mL鹽酸、2.5mL硝酸,加熱至巖石樣品完全溶解,隨后加入5mL氫氟酸、0.5mL高氯酸,將燒杯置于電熱板上加熱分解,蒸發至燒杯中的白煙冒盡,取下,用少量的蒸餾水沖洗燒杯內壁,加入2mL濃鹽酸,微熱溶解樣品溶液中的鹽類,取下冷卻,移入25mL容量瓶中,加水稀釋至刻度線處,搖勻。
三、結果與討論
1、樣品測定條件
本次實驗中樣品的溶解溫度設定為180℃,根據各種巖石的特點,選擇不同的溶樣時間,以促進樣品的完全溶解。對于本次實驗中選取的巖石來說,在180℃的條件下溶樣24-40h就可以使巖石樣本完全溶解。花崗巖中含有較多的難溶的礦物成分,因此其溶樣時間較長,通常需要40h左右才可以完全溶解,而一般的鉛礦石和鋅礦石較易溶解,一般在24h左右就可以完全溶解。
2、方法的準確度與精密度
根據樣品的分析步驟同步操作以上各巖石,利用ICP-MS進行測定,其標準偏差見表3,統計的數據結果顯示,其精密度(RSD)和準確度(RE)都在10%的范圍內,大多數的元素精密度和準確度在5%范圍內。
3、微量元素的不確定度評定
3.1 樣品稱量過程中產生的不確定度
3.1.1 天平校準產生的不確定度
由天平的檢定可以得知,在天平0-5g的量程范圍內,天平允許的誤差是±0.01 mg,由矩形分布的規律可知,,天平校準產生的不確定度為:

3.1.2 重復稱量的不確定度
為了獲得較為可靠的重復性稱量的不確定度,在電子天平上稱取0.12500g的試樣,同時進行10次的重復稱量,計算得出的質量平均值為0.12503g,通過貝塞爾公式可以計算出標準偏差:

對同一試樣進行反復測量而導致的不確定度為:

3.1.3 稱量的合成標準不確定度
將上述兩方面得到的兩項標準偏差進行合成,可以得出稱量的合成標準不確定度為:

稱量的試樣質量為0.12500g,所以相對的標準不確定度為:

3.2 濃度測量過程中產生的不確定度
3.2.1 標準溶液的不確定度
(1)標準儲備液的不確定度
在多元素的混合標準溶液(ρ1=1000ug/mL)中,這一溶液濃度的允許差值為±0.5%,依照矩形分布評定,可以計算出標準儲備液的不確定度為:

進一步得出其相對不確定度為:
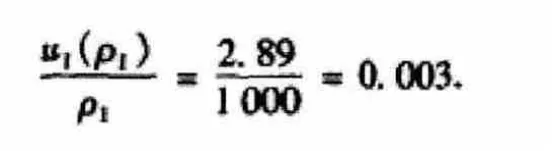
(2)標準溶液稀釋配制過程中引起的不確定度


3.2.2 重復性實驗產生的不確定度
為了較為準確地得到重復性測量的不準確度,對樣品溶液可以進行5~10次的重復測量,通過測量巖石中各元素含量的平均值,并計算其標準偏差和相對標準不確定度,就可以計算出由重復性實驗而導致的不確定度。
3.3 合成不確定度


表2 各組分不確定度分量及結果的表達
結語
本文采用混合酸的方法分解巖石樣品,并利用等離子光譜法測定巖石中各微量元素的相對含量,這一方法簡單快速,能夠同時測定多個元素,并且,在此次測定的元素中,大多數的元素精密度和準確度保持在5%的范圍內,結果準確可靠,且分析出了實驗中各元素的不確定度,為今后巖石中各元素的測定和分析提供了一定的依據和參考。
[1]楊曉紅,張林群,周愛東,等.等離子光譜法測定巖石中微量元素的不確定度評定[J].南京師大學報.2011(3):69-73.
[2]楊春茹,康俊,高建文,等.電感耦合等離子體質譜法測定煤中多種微量元素[J].煤質技術.2013(4):1-4.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
