異基因造血干細胞移植治療骨髓增生異常綜合征合并壞疽性膿皮病一例
2015-07-07 10:07:12吳婷婷王曉燕吳迪炯俞慶宏邵科釘沈一平周郁鴻葉寶東
中華移植雜志(電子版) 2015年3期
吳婷婷 王曉燕 吳迪炯 俞慶宏 邵科釘 沈一平 周郁鴻 葉寶東
骨髓增生異常綜合征(myelodysplastic syndromes,MDS)是一組異質性克隆性造血干細胞疾病,其病因尚未確定。異基因造血干細胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT)是目前治療MDS 的有效方法,尤其是對于青年患者而言[1]。壞疽性膿皮病(pyoderma gangrenosum,PG)是一種以皮膚破壞性潰瘍為特征的反應性炎癥性皮膚病,較為少見,目前認為可能是一種免疫介導性疾病,臨床上可能伴發炎癥性腸病、MDS 等。目前臨床上主要應用糖皮質激素和免疫抑制劑治療PG[2]。關于allo-HSCT 治療MDS 合并PG 的報道較少見,浙江中醫藥大學附屬第一醫院血液科應用allo-HSCT 治愈了1 例MDS 合并PG 患者,現報道如下。
1 臨床資料
患者男性,19 歲,既往體健。2009 年10 月因“頭暈、面黃、乏力伴發熱4 d”到當地醫院就診,查血常規:白細胞1.2 ×109/L,血紅蛋白117 g/L,血小板52 ×109/L;當時未進一步檢查明確診斷,給予中成藥、雄激素口服,效果欠佳。2009 年12 月到我院就診收治入院,患者否認工作粉塵、毒物、放射性物質接觸史;家中父母及姐妹均體健,否認家族遺傳病史。查血常規示:白細胞2.0 ×109/L,血紅蛋白71 g/L,血小板96 × 109/L;網織紅細胞計數1.87%。鐵蛋白238.1 ng/mL,葉酸47 nmol/L,維生素B12 723 pmol/L。骨髓涂片檢查示:骨髓小粒可見,骨髓增生活躍,其中粒系增生活躍,占32.5%,各階段粒細胞比例、形態大致正常;紅系增生活躍,占45.5%,各階段紅細胞比例大致正常,可見部分紅系細胞類巨幼樣變,核漿成熟失衡,紅細胞呈卵圓形;淋巴細胞占20%,形態大致正常,全片見巨核細胞78 個,可見小巨核細胞,提示MDS-難治性貧血(refractory anemia,RA)型。骨髓活檢:骨髓部分區域增生極度低下,部分區域增生較活躍,粒紅比例減小,粒紅系細胞以偏成熟為主,可見幼稚前體細胞異常定位現象,Gomori 網狀纖維染色(-)。染色體檢查示:46XY,正常核型。腹部B 超示脾臟形態、大小正常,厚3.4 cm。臨床診斷為MDS-RA,國際預后積分為中危-1。給予十一酸睪酮、沙利度胺、曲安西龍口服,注射氨磷汀以及輸注血制品等對癥治療,患者病情反復,期間多次發生皮膚淤點淤斑、感染,予卡絡磺鈉止血、抗炎治療后好轉出院。
2010 年7 月,患者無明顯誘因出現右下肢近膝端紅腫、疼痛,逐漸化膿潰爛,時有發熱,最高體溫達40 ℃。門診就診后收治入院,給予廣譜抗生素治療,患者癥狀無緩解,右下肢皮膚潰瘍面不斷向周圍擴展,直徑達10 cm 以上,潰瘍較深且伴組織壞死。局部皮膚清創同時行病理活檢,潰瘍組織病理示:表皮輕度角化過度,鱗狀上皮增生,灶性潰瘍形成,真皮水腫,纖維細胞增生,小血管增生、擴張、出血,可見數個上皮樣結節,伴多核巨細胞反應;見數個裂隙樣潰瘍,較多中性粒細胞、組織細胞浸潤;散在含鐵血黃素沉著(見圖1);考慮為PG。潰瘍分泌物細菌、真菌培養均未找到致病菌。后加用甲潑尼龍、環孢素和沙利度胺治療,患者體溫逐漸恢復正常,皮膚潰瘍趨于收斂,患者疼痛好轉后出院。出院2 周門診隨訪發現,患者血紅蛋白持續下降,最低時達20 g/L,完全依賴輸注紅細胞維持生命;且右下肢近膝端處PG 又反復發作2 次,加大甲潑尼龍劑量(40 mg/d)后好轉。
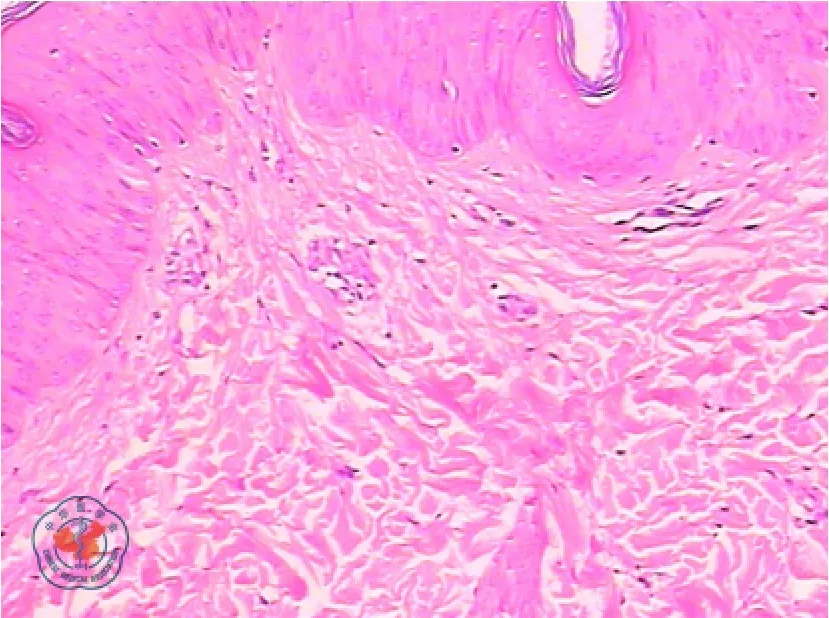
圖1 2010 年7 月骨髓增生異常綜合征合并壞疽性膿皮病患者右下肢近膝端潰瘍組織病理檢查結果(HE ×400)
2011 年8 月,患者考慮行allo-HSCT 再次入住我院。供者為其胞姐,HLA 配型全相合(6/6),ABO 血型相合。入院實驗室檢查:白細胞0.9 ×109/L,血紅蛋白27 g/L,血小板30×109/L;鐵蛋白3 859.1 ng/mL;骨髓涂片檢查仍提示MDS-RA。2011 年9 月4 日,患者右側手臂上方近三角肌處出現一類圓形皮損,行局部組織活檢,仍提示PG 可能。2011 年9 月6 日起予改良BU/CY 方案預處理,-8、-7、-6 d 給予馬利蘭4 mg·kg-1·d-1,-5、-4 d 給予環磷酰胺1.8 g·(m2)-1·d-1。移植物抗宿主病(graftversus-host disease,GVHD)預防方案為環孢素和短程甲氨蝶呤,從-5 d 開始靜脈滴注環孢素1.5 ~3.0 mg·kg-1·d-1,待患者無明顯不良反應改為口服6 ~8 mg·kg-1·d-1,分2 次服用,再根據其血藥濃度谷值(維持在150 ~250 μg/L)酌情減量至1.5 ~2.0 mg·kg-1·d-1,共用3 ~6 個月;移植后+1 d予甲氨蝶呤15 mg/m2,+3、+6、+11 d 分別予10 mg/m2。回輸其胞姐外周血CD34+細胞為4.72 ×106/kg,移植過程順利。移植12 d 患者造血功能重建,新出現的皮損消失。移植后1 個月通過短串聯重復序列PCR 檢測證實造血干細胞完全植入。移植過程中和移植后患者均未出現其他移植相關并發癥,移植后10 個月停用免疫抑制劑。移植后1 年查血常規示:白細胞4. 7 × 109/L,血紅蛋白136 g/L,血小板132 ×109/L。骨髓涂片檢查示:骨髓小粒可見,骨髓增生活躍,其中粒系增生尚活躍,占31.5%,各階段粒細胞比例略偏低,形態大致正常;紅系增生明顯活躍,占57%,以中晚幼紅細胞為主,形態未見明顯異常;淋巴細胞占8.5%,形態正常;全片見巨核細胞104 個,其中產板巨核細胞占24%,血小板呈中小簇分布,數量大致正常。截至2014 年9 月,該患者已隨訪3 年,隨訪期間復查血常規均在正常范圍,未再次復查骨髓象,未觀察到類似皮損出現,考慮MDS 和PG 均無復發。
2 討 論
MDS 是起源于造血干細胞的一組異質性髓系克隆性疾病,部分患者會轉變為急性髓細胞白血病。年輕MDS 患者(<50 歲)臨床上較少見,與年老MDS 患者相比,年輕患者疾病特點是女性多于男性,臨床癥狀較輕,嚴重貧血更少見,腎功能損害程度較輕,骨髓中的原始細胞比例更少,RA 和單系造血細胞異常類型的患者比例更高,其總生存期明顯高于年老MDS 患者[3]。但是,年輕MDS 患者疾病進展過程與年老患者相似,二者發展為急性髓細胞白血病的概率是一樣的,提示臨床表現相似的MDS患者中,年輕MDS 患者具有更高風險和較差預后[3]。盡管近年來我們對MDS 的分子和遺傳學特征有了更深刻的認識,但是唯一可能治愈MDS 的治療方法仍然是應用allo-HSCT[1],尤其是對于年輕MDS 患者而言。
PG 是一種非感染性的、以中性粒細胞浸潤為特征的潰瘍性皮膚疾病,臨床上較罕見,但病情較嚴重,多與一些系統性疾病有關。其發病機制尚不明確,現多認為免疫機制在PG 的發生中起了主導作用。超過50%的PG 患者合并系統性疾病,包括潰瘍性結腸炎、MDS、克羅恩病和類風濕性關節炎等[2]。Su 等[4]于2004 年提出的PG 診斷標準是目前我們診斷PG 的主要依據,主要根據臨床表現、組織病理學和基礎疾病等診斷。PG 的治療主要是應用免疫抑制劑控制病情,包括環孢素、皮質類固醇、氨苯砜、米諾環素和沙利度胺等。
對于MDS 合并PG 的患者,往往應用皮質類固醇聯合沙利度胺進行治療[5-6];其中部分對口服類固醇無效的患者,靜脈應用甲潑尼龍可以提高療效[5]。也有報道應用大劑量丙種球蛋白治療MDS合并PG 的患者取得了較好的療效[7]。Allo-HSCT在理論上可完全或接近完全免疫重建,是治愈自身免疫性疾病的理想方式,因此有些移植中心開始應用allo-HSCT 治療難治性自身免疫性疾病[8],但由于GVHD 等移植相關并發癥發生率較高,限制了其在難治性自身免疫性疾病中的推廣應用。PG 患者的長期生存仍是不可預測的,最初對藥物治療有反應的PG 患者中絕大多數會復發[2],應用潑尼松和環孢素治療的患者疾病復發的比例分別為70%和66%[9],有一些類型的PG 病死率高達30%[10]。我們曾報道過1 例MDS-RA 合并PG 患者,給予潑尼松、嗎替麥考酚酯和沙利度胺治療后病情曾一度控制[11],但最終在治療19 個月后死亡。
MDS 合并PG 患者應用allo-HSCT 治療的臨床病例目前比較少見。本例患者經過骨髓涂片檢查、骨髓活檢和染色體檢查等明確診斷為MDS,在治療MDS 的過程中,無明顯誘因出現了快速進展的疼痛性潰瘍、高熱,結合組織病理結果、細菌和真菌培養陰性以及多種抗生素治療無效等表現,綜合考慮為PG,在診斷上是比較明確的。在給予甲潑尼龍、環孢素和沙利度胺聯合治療后,患者病情一度得到緩解,但是PG 復發2 次,且MDS 病情持續進展,期間一度完全依賴輸注血制品維持生命。行allo-HSCT 10 個月后患者停用免疫抑制劑,目前已經隨訪3年,患者MDS 和PG 均未復發。根據這例患者的診治經過結合相關文獻,我們建議,對于MDS 合并PG的年輕患者應盡早考慮行allo-HSCT,以完全控制MDS 和PG。
1 Vaughn JE,Scott BL,Deeg HJ. Transplantation for myelodysplastic syndromes 2013[J]. Curr Opin Hematol,2013,20(6):494-500.
2 Wollina U. Pyoderma gangrenosum—a review[J]. Orphanet J Rare Dis,2007,2:19.
3 Marisavljevic D,Savic A,Zeremski V,et al. Myelodysplastic syndromes in adults aged less than 50 years: incidence and clinicopathological data[J]. J BUON,2014,19(4):999-1005.
4 Su WP,Davis MD,Weenig RH,et al. Pyoderma gangrenosum:clinicopathologic correlation and proposed diagnostic criteria[J]. Int J Dermatol,2004,43(11):790-800.
5 Yamauchi T,Ishida K,Iwashima Y,et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome[J]. J Infect Chemother,2003,9(3):268-271.
6 Koca E,Duman AE,Cetiner D,et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum[J]. Neth J Med,2006,64(11):422-424.
7 Tamaki K,Nakazawa T,Mamehara A,et al. Successful treatment of pyoderma gangrenosum associated with myelodysplastic syndrome using high-dose intravenous immunoglobulin[J]. Inter Med,2008,47(23):2077-2081.
8 Strober J,Cowan MJ,Horn BN. Allogeneic hematopoietic cell transplantation for refractory myasthenia gravis[J]. Arch Neurol,2009,66(5):659-661.
9 Vidal D,Puig L,Gilaberte M,et al. Review of 26 cases of classical pyoderma gangrenosum:clinical and therapeutic features[J]. J Dermatolog Treat,2004,15(3):146-152.
10 Hafner J,Kühne A,Trüeb RM. Successful grafting with EpiDex in pyoderma gangrenosum[J]. Dermatology,2006,212(3):258-259.
11 金玥,俞慶宏,周郁鴻,等. 骨髓增生異常綜合征伴壞疽性膿皮病1 例[J]. 臨床血液學雜志,2008,21(5):498-499.
