基因組高通量測序數(shù)據(jù)結(jié)構(gòu)變異識別算法
2015-04-29 00:44:03王春宇等
智能計算機與應(yīng)用 2015年1期
王春宇等
摘 要:遺傳變異是生命的基本特征,遺傳變異與表型差異之間的關(guān)系,是現(xiàn)代生物學(xué)的一個基本問題。由基因決定生物體的遺傳特征和主要個體差異的觀念正在逐漸改變,過去幾年的許多研究顯示,基因組中大尺度的結(jié)構(gòu)變異與個體的表型差異和疾病等有一定的關(guān)聯(lián)。有關(guān)遺傳變異和表型多樣性的研究,需要比較生物體個體基因組間的各種不同。利用NGS數(shù)據(jù)全面分析結(jié)構(gòu)變異的技術(shù)目前仍然不成熟。因此本文根據(jù)生物學(xué)知識,利用高通量測序數(shù)據(jù),對植物基因組結(jié)構(gòu)變異的識別問題深入系統(tǒng)的研究,提出新的結(jié)構(gòu)變異識別方法和精確的斷點預(yù)測方法。
關(guān)鍵詞:高通量測序;遺傳變異;結(jié)構(gòu)變異;斷點預(yù)測
中圖法分類號:TP391 文獻標(biāo)識碼:A 文章編號:2095-2163(2015)01-
Abstract: Structural variations are genomic rearrangements that contribute significantly to evolution, natural variation between organisms, and are often involved in biological phenotypes and genetic disorders. Traditional microscope and array based methods are used for the detection of larger events or copy number variations. Next generation sequencing has enabled the detection of all types of structural variants from genome accurately. In practice, a significant challenge lies in the development of computational methods that are able to identify these structural variants based on the generated high-throughput sequencing data. In this project, the paper focuses on the design and implementation of the algorithms for multiple donor plant genomic structural variation identification with the combination of assembly, pair-end mapping, split read and depth of coverage analyzing.
Keywords: High-Throughput Sequencing; Genetic Variation; Structural Variation; Breakpoint Prediction
0 引 言
遺傳變異是生命的基本特征,遺傳變異與表型差異之間的關(guān)系,是現(xiàn)代生物學(xué)的一個基本問題。由基因決定生物體的遺傳特征和主要個體差異的觀念正在逐漸改變,過去幾年的許多研究顯示,基因組中大尺度的結(jié)構(gòu)變異(structural variation, SV)與個體的表型差異和疾病等有一定的關(guān)聯(lián)[1]。近年的研究更進一步,很多疾病和表型差異與基因組的結(jié)構(gòu)變異有關(guān)系,人們已經(jīng)逐漸認(rèn)識到結(jié)構(gòu)變異的普遍性和重要性[2-3]。
1 結(jié)構(gòu)變異及其重要作用
基因組結(jié)構(gòu)變異通常指基因組DNA分子上整片的片段差異,包括保持?jǐn)?shù)量平衡的倒位(inversion)和易位(translocation)變化以及導(dǎo)致數(shù)量失衡的插入(insertion)、缺失(deletion)和重復(fù)(duplication)變化,其中數(shù)量失衡的變化也稱為拷貝數(shù)變異(copy number variation, CNV)。一般認(rèn)為結(jié)構(gòu)變異的片段,長度約在一千到幾百萬個堿基之間,一致性在90%以上。2004年,全球范圍內(nèi)數(shù)個“人類基因組計劃”研究基地意外地發(fā)現(xiàn),表型正常的人群中,不同個體間在某些基因的拷貝數(shù)上存在差異,結(jié)構(gòu)變異的發(fā)現(xiàn)及其對基因雜合性的重要作用,徹底改變了人們對基因型研究的認(rèn)識[5]。
個體基因組之間的差異,除少數(shù)SNP和短序列的插入/缺失(indel)變化之外,大部分以結(jié)構(gòu)變異為主[6]。相關(guān)研究也證實,在玉米自交系中存在異常高的結(jié)構(gòu)變異[7]。雖然普遍認(rèn)為當(dāng)基因組規(guī)模相對較小時,發(fā)生結(jié)構(gòu)變異的數(shù)量會較少,但對大豆的研究卻特別發(fā)現(xiàn),相對于大豆基因組的大小和其中SNP的數(shù)量來說,結(jié)構(gòu)變異的發(fā)生率較高[8]。而且結(jié)構(gòu)變異在大豆的不同染色體間的分布也不均勻,在基因分布較密的區(qū)域發(fā)生較多,并且與植物的抗性表現(xiàn)有一定的關(guān)聯(lián)[9]。
2 相關(guān)研究
最近幾年隨著下一代測序技術(shù)中高通量測序平臺在應(yīng)用上的推廣普及,基因組序列數(shù)據(jù)的獲取速度越來越快。這些平臺在測序中都會產(chǎn)生大量的相互交疊的對端讀片段(paired-end read)。被測序的DNA分子將打斷為很多碎片,其插入長度約2k~150kbp,設(shè)備僅測每個碎片兩端長度為25~1kbp的對端讀片段。使用這樣的序列數(shù)據(jù)識別結(jié)構(gòu)變異,在長度和精確度上均會高于芯片技術(shù)的使用效果[10],因而適用于各種類型結(jié)構(gòu)變異的識別以及新類型的發(fā)現(xiàn)。采用測序技術(shù)的結(jié)構(gòu)變異識別可以使用重頭組裝和重測序兩種方法。其中,重頭組裝方法最為理想,但由于目前測序技術(shù)的限制,完整的全基因組組裝結(jié)果并非最優(yōu),不能很好地滿足先組裝、再識別的要求;而重測序方法則更加實際,可將得到的大量讀片段定位到參考基因組并實施比較分析。讀片段定位(read mapping)是序列比對的基本任務(wù),對其研究已可堪稱深入,但通用的讀序列定位算法應(yīng)用于結(jié)構(gòu)變異檢測時,卻沒有充分考慮斷點(breakpoint)處的比對,從而會導(dǎo)致斷點預(yù)測不準(zhǔn)確甚至結(jié)構(gòu)變異檢測失敗[11]。
采用重測序技術(shù)的結(jié)構(gòu)變異分析方法主要有三種,具體來說,分別是:
(1)覆蓋度(read depth或depth of coverage, DoC)分析法,根據(jù)在參考基因組上讀片段的覆蓋度,判斷是否有拷貝數(shù)變化的結(jié)構(gòu)變異,如CNV-seq[12]等,這類方法無法識別拷貝數(shù)不變的倒位和易位變化;
(2)對端定位(paired-end mapping, PEM)法,根據(jù)對端讀片段插入距離的一致性,判斷是否發(fā)生結(jié)構(gòu)變異,如VariationHunter等;
(3)讀片段分割法(split read),利用讀片段在參考基因組上定位時的斷點判斷結(jié)構(gòu)變異,如Pindel[13],但由于NGS測序的讀長較短,不利于發(fā)現(xiàn)較大的結(jié)構(gòu)變異。也有將這些信息結(jié)合的方法,如CNVer[14]等只關(guān)注了CNV一種結(jié)構(gòu)變異;Break- Dancer[15]和GASV[16]使用了高置信度讀片段,忽略了與重復(fù)區(qū)域有關(guān)的低置信度讀片段;inGAP-sv和Gnome Strip[17]在精確性和特異性兩方面都較好,但卻都基于二倍體人類基因組設(shè)計和實現(xiàn)。因此,目前采用NGS數(shù)據(jù)進行基因組結(jié)構(gòu)變異的檢測與分析方法還不夠成熟,缺少完整的分析、發(fā)現(xiàn)和評價體系。
3 結(jié)構(gòu)變異識別方法
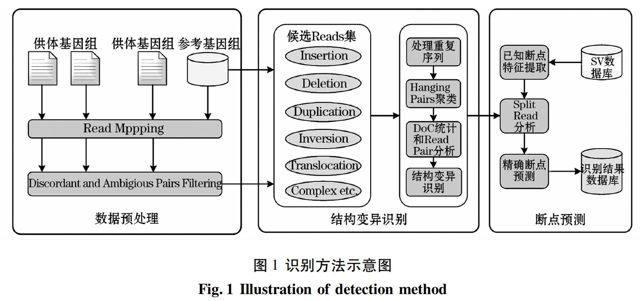
本文的基本的結(jié)構(gòu)變異識別方法如圖1所示,其原理可概括為:以目標(biāo)物種基因組序列為參考,使用多個供體全基因組高通量末端配對測序讀片段數(shù)據(jù),采用PEM的方法識別結(jié)構(gòu)變異,并利用對端信息進行讀片段分隔方式的斷點預(yù)測。本文設(shè)計基于單個參考基因組和多供體基因組重測序數(shù)據(jù)的結(jié)構(gòu)變異識別方法。從識別方法的角度,本文可以分為三個主要方面:數(shù)據(jù)預(yù)處理,結(jié)構(gòu)變異識別和精確的斷點預(yù)測。下面將給出其具體實現(xiàn)。
3.1 數(shù)據(jù)預(yù)處理
數(shù)據(jù)預(yù)處理的主要任務(wù)是讀片段定位,不一致和歧義讀對(discordant and ambiguous pairs)的過濾,產(chǎn)生的中間結(jié)果為不同類型的候選讀片段集,用于支持可能的多種結(jié)構(gòu)變異的識別和斷點精確預(yù)測。在此,對其過程解析可做如下展現(xiàn)。
(1)讀片段定位
在參考基因組上定位讀片段,對每個供體的讀片段分別向參考基因組上定位,相應(yīng)得到各自的定位結(jié)果。定位時除需考慮一般定位算法涉及的測序錯誤、soft clipping等問題之外,還需要考慮兩方面問題。具體描述為:
① 多處匹配:若同一個讀片段可能在多處得到較好的匹配,則可能因為有拷貝數(shù)變化,需采用基于種子及擴展的比對算法,保存并返回多個高分比對位置;
② 對讀片段分割的懲罰:若某條讀片段剛好在斷點處,由于對空位的罰分,可能造成錯誤定位或無法定位,因此要調(diào)整比對算法的罰分策略,允許一處不罰分的長空格。
(2)不一致讀片段對的過濾
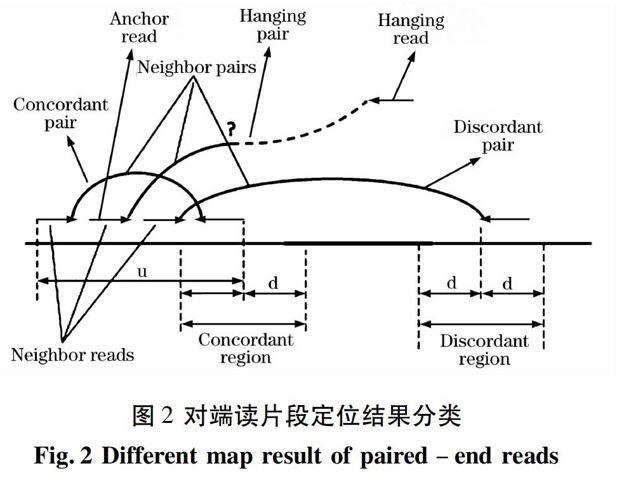
由測序技術(shù)可知,同一碎片雙端測序的讀片段對,在參考基因組上的定位如若符合一定的方向和距離要求即稱為一致的(concordant),否則稱為不一致的(discordant),如圖2所示。不一致的原因之一就是由于參考基因組和供體基因組之間的結(jié)構(gòu)差異,主要分為三類,將其綜合概述如下:
①方向異常的,可能因為倒位,利用定位的方向可篩選出;
② 距離異常,可能對應(yīng)插入、缺失或易位,根據(jù)測序參數(shù)中平均插入長度L和標(biāo)準(zhǔn)差σ來處理,當(dāng)距離超過L+3σ錯誤!未指定書簽。和小于L-3σ錯誤!未指定書簽。的都視為異常并篩選出;
③ 懸掛對(hanging pairs),即對端讀片段中有一條未能成功定位的,可能對應(yīng)較大的結(jié)構(gòu)變異,由定位結(jié)果可直接篩選出來。
(3)歧義讀片段對過濾
若一條讀片段在多處均有較好定位,又可將其稱為歧義定位對。可能由于潛在的結(jié)構(gòu)變異造成,同樣按照類似不一致讀對過濾的方法進行篩選。只是要依據(jù)在第(1)步中讀片段定位算法所提供的結(jié)果,即將歧義讀片段對的多處匹配結(jié)果統(tǒng)一、完整地實現(xiàn)返回。
(4)合并多供體的候選讀片段
由于文庫制備和測序?qū)嶒灥牟煌總€供體測序的基本參數(shù)也是不同的,如插入長度、讀片段長度等,所以讀片段的定位和過濾即需分別設(shè)計運行。而讀片段的定位結(jié)果參考同一基因組,因此可將過濾得到的候選讀片段按類型合并,并保存供體來源信息,并在發(fā)生分歧時提供分類依據(jù)。而相應(yīng)于懸掛對,則由于只有一條讀片段能夠成功定位,需要對未成功定位的讀片段連同歧義讀片段進行聚類,以發(fā)現(xiàn)較大的結(jié)構(gòu)變異。
3.2 結(jié)構(gòu)變異識別
(1)基本結(jié)構(gòu)變異識別
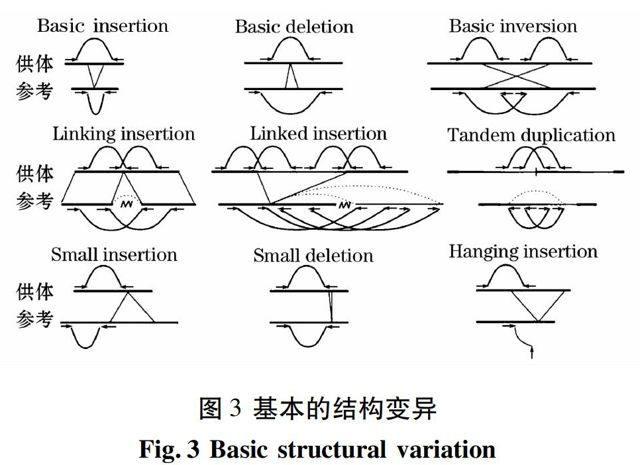
由結(jié)構(gòu)變異導(dǎo)致的不一致讀對如圖3所示。對于基本的插入、缺失和倒位等,需根據(jù)定位的座位進行分類并計算讀對共同的比對得分。對每個座位,若有3組以上來自同一供體的同類讀對,則認(rèn)為是合法的結(jié)構(gòu)變異;或者,另有多個供體且每個有2組以上的同類讀對,也將認(rèn)定為是合法的結(jié)構(gòu)變異。
(2)讀片段覆蓋度分析
利用多供體數(shù)據(jù),在提高等效的覆蓋度方面具有顯著優(yōu)勢。覆蓋度分析的原理如圖4所示,當(dāng)覆蓋度發(fā)生異常時,可能因為基因組的重復(fù)片段或存在較大的結(jié)構(gòu)變異。分析定位到參考基因組上讀片段的數(shù)量,就需要估計全部成功定位的讀片段在參考基因組上的分布和特征,再根據(jù)總體及局部的分布以判斷是否呈現(xiàn)有明顯的變化,并在一定大小的窗口內(nèi)計算讀片段覆蓋度,而當(dāng)信號發(fā)生相對顯著的增加和減少時,即可判斷有結(jié)構(gòu)變異發(fā)生。
覆蓋度與對端讀片段信息的結(jié)合分析,可發(fā)現(xiàn)較大結(jié)構(gòu)變異的原理如圖5所示。具體地,由于幾個對端讀片段的距離較大并且同時此處的覆蓋度超過正常值。在覆蓋度異常的區(qū)域,尋找方向不一致的對端讀片段,并試圖利用復(fù)制該區(qū)域的方法,將不一致的對端讀片段一致排列,以發(fā)現(xiàn)結(jié)構(gòu)變異。
(3) 復(fù)雜結(jié)構(gòu)變異識別與重復(fù)片段的分析
對于超過插入長度的結(jié)構(gòu)變異,會導(dǎo)致懸掛對和歧義定位。因此對這些讀片段,使用貪心算法將距離和方向類似的片段實現(xiàn)聚類后,即需進行基于de Bruijn圖的拼接。在此圖中對數(shù)據(jù)錯誤和重復(fù)序列進行處理,并根據(jù)路徑與參考基因組實行比較,從而分析可能存在的結(jié)構(gòu)變異。如圖6所示,利用多供體信息,可以相互驗證路徑中的環(huán)路,是否是因為重復(fù)片段。若構(gòu)成路徑的片段相對供體來源是均勻的,可以認(rèn)為是在供體中普遍存在的重復(fù)序列;否則即認(rèn)為是由于結(jié)構(gòu)變異,再與參考基因組相比較,并確認(rèn)結(jié)構(gòu)變異的類型和位置。
3.3 結(jié)構(gòu)變異的斷點預(yù)測
斷點預(yù)測需要讀片段分割分析和對端讀片段分析相結(jié)合,來自多供體的對端讀片段構(gòu)成差異較大,更利于斷點位置的精確預(yù)測。如果有多個讀片段在斷點處的一側(cè)發(fā)生完美匹配,則可以通過該組讀片段直接預(yù)測斷點位置,誤差在讀片段間交疊的長度范圍內(nèi);而如果在讀片段覆蓋范圍之外,則需要使用對端讀片段的插入長度進行分析。因為插入長度L有一定誤差范圍,假設(shè)在Lmin和Lmax之間且對端讀片段的左右斷點分別是(x, y),那么可以估計斷點發(fā)生的位置(a, b)滿足 Lmin ≤ (a-x) + (y-b) ≤ Lmax,即(a, b)落在x, y, Lmin, Lmax構(gòu)成的梯形區(qū)域當(dāng)中。若有多個對端讀片段同時支持該區(qū)域的結(jié)構(gòu)變異,則該斷點所在位置需要在多個梯形的交疊區(qū)域中,如圖7中分別為形成插入和缺失時的情況。此外,為了能夠處理超過插入長度結(jié)構(gòu)變異的斷點,利用斷點兩翼序列的特點,根據(jù)斷點處的微同源性,分析已知結(jié)構(gòu)變異斷點處的微同源特征和兩翼序列的特征,再進行斷點預(yù)測的。
4 實驗結(jié)果及分析
本文實驗利用模擬數(shù)據(jù)生成供體的測序片段,根據(jù)dbVar對已知結(jié)構(gòu)變異大小和分布,在大豆1號染色體上構(gòu)造了插入和缺失類型的結(jié)構(gòu)變異,如表1所示。由于易位、倒位和重復(fù)事件現(xiàn)有數(shù)據(jù)中也很少,而且通常各種軟件對這些事件識別的準(zhǔn)確率都不高,所以沒有進行模擬。本文即比較選擇了主流的幾個結(jié)構(gòu)變異識別工具,分別是BreakDancer、Pindel和CREST。
對缺失事件的識別結(jié)果如表2所示。綜合該結(jié)果可知,總體而言,Pindel、BreakDancer和本文方法的識別結(jié)果各有優(yōu)勢,不可概述;分別地,Pindel和本文方法在小型的缺失事件上結(jié)果較好,而在100bp以上的范圍SVdetect、BreakDancer和本文方法的結(jié)果則更佳。其中100~1000bp的兩組結(jié)果中,本文方法識別效果較好,而在100~500bp和 > 1 000b兩組結(jié)果中當(dāng)屬本文方法的效能最優(yōu)。主要原因是對于利用split-read方式的識別工具,其大小產(chǎn)生split-read的數(shù)量較少,而對于不一致讀片段對的識別工具其長度卻恰好落入插入長度的誤差范圍內(nèi)。
綜合分析實驗結(jié)果,本文方法在不同的條件下均可以得出較好的識別結(jié)果。本文方法能夠準(zhǔn)確識別出大部分的結(jié)構(gòu)變異事件,因而可以用于NGS數(shù)據(jù)中結(jié)構(gòu)變異的識別。
參考文獻:
[1] CRADDOCK N, Wellcome Trust Case Control Consortium, CRADDOCK N, HURLES ME, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls[J]. Nature, 2010, 464(7289): 713–20.
[2] ABECASIS G R, ALTSHULER D, AUTON A, et al. A map of human genome variation from population-scale sequencing[J]. Nature, 2010, 467(7319):1061–73.
[3] 周雪崖, 張學(xué)工. 基于拷貝數(shù)變異的遺傳關(guān)聯(lián)研究[J]. 科學(xué)通報, 2011, 56(6):370-182.
[4] TREANGEN T J, SALZBERG S L. Repetitive DNA and next-generation sequencing: computational challenges and solutions[J]. Nature reviews. Genetics, 2012, 13(1):36–46.
[5] FEUK L, CARSON A R, SCHERER S W. Structural variation in the human genome[J]. Nature reviews. Genetics, 2006, 7(2):85–97.
[6] Le ROUZIC A, BOUTIN T S, CAPY P. Long-term evolution of transposable elements[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104(49):19375–80.
[7] SPRINGER N M, YING K, FU Y, et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content[J]. PLoS genetics, 2009, 5(11): e1000734.
[8] HAUN W J, HYTEN D L, XU W W, et al. The composition and origins of genomic variation among individuals of the soybean reference cultivar Williams 82[J]. Plant physiology, 2011, 155(2):645–655.
[9] MCHALE L K, HAUN W J, XU W W, et al. Structural variants in the soybean genome localize to clusters of biotic stress-response genes[J]. Plant physiology, 2012, 159(4):1295–1308.
[10] QI J, ZHAO F. inGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data[J]. Nucleic acids research, 2011, 39(Web Server issue):W567–575.
[11] TEO S M, PAWITAN Y, KU C S, et al. Statistical challenges associated with detecting copy number variations with next-generation sequencing[J]. Bioinformatics (Oxford, England), 2012, 28(21):2711–2718.
[12] XIE C, TAMMI M T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing.,” BMC bioinformatics, 2009, 10:80.
[13] YE K, SCHULZ M H, LONG Q, et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads[J]. Bioinformatics (Oxford, England), 2009, 25(21):2865–71.
[14] MEDVEDEV P, FIUME M, DZAMBA M, et al. Detecting copy number variation with mated short reads[J]. Genome research, 2010, 20(11):1613–22.
[15] CHEN K, WALLIS J W, MCLELLAN M D, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation[J]. Nature methods, 2009, 6(9):677–681.
[16] SINDI S, HELMAN E, BASHIR A, et al. A geometric approach for classification and comparison of structural variants[J]. Bioinformatics (Oxford, England), 2009, 25(12):i222–230.
[17] HANDSAKER R E, KORN J M, NEMESH J, et al. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale[J]. Nature genetics, 2011, 43(3):269–276.
