層狀M oS2/Graphene薄膜的制備及其電催化制氫性能
2015-03-19 08:25:22楊陽許頔吳卿永項民刁鵬
北京航空航天大學學報 2015年11期
關鍵詞:催化劑
楊陽,許頔,吳卿永,項民,刁鵬
(北京航空航天大學 材料科學與工程學院,北京 100191)
氫氣的燃燒產物為水,不會對環境造成任何污染,其可以作為清潔能源應用在生活的各個方面[1-3].因此,在能源和環境危機越來越嚴重的今天,氫能受到了大家的高度重視.氫氣最豐富的來源為水,尋找高效的分解水制氫的催化劑迫在眉睫.目前電催化制氫效率最高的催化劑為Pt族金屬[4-5],但其嚴重受限于自然稀缺性和高昂成本,因此急需開發出能替代Pt族金屬的經濟、穩定、環保、高效的新型催化劑.
MoS2作為一種制氫的電催化劑,因其產量豐富、價格便宜而得到了廣泛的關注.理論和實驗[6-7]證實,MoS2的電催化制氫的活性位點主要在其邊緣位置[8-9].因此,制備納米級的 MoS2,使其更多的邊緣暴露在外面,是提高其電催化制氫活性的有效途徑.但是納米級的MoS2很容易團聚,而以某種導電材料為基底,就有可能合成出層狀的、具有豐富邊緣的MoS2電催化劑.如文獻[8,10-13]中所報道的以 Au[8,12]、碳納米管[13]等為基體,成功地制備出了具有高效催化活性的層狀的MoS2復合電催化劑.
本實驗中采用水熱合成的方法,以二水合鉬酸 鈉 (Na2MoO4·2H2O)為 前 驅 體,硫 脲(NH2CSNH2)為硫源和還原劑,合成了層狀的MoS2/Graphene電催化劑.MoS2通過化學耦合選擇性地生長在石墨烯上,使得MoS2邊緣擁有豐富的活性位點,石墨烯作為良好的導電基體也能大大加快電子由層狀邊緣的活性位點轉移到溶液中的速度.因此,MoS2/Graphene表現出了良好的電催化制氫性能,其作為Pt族貴金屬的替代品,具有廣闊的應用前景.
1 實驗部分
1.1 化學試劑與材料
Na2MoO4·2H2O(阿法埃莎,99.99%),NH2CSNH2(阿法埃莎,99%),石墨薄片(阿法埃莎,99.8%),高錳酸鉀(KMnO4,北京化工廠,AR(分析純)),濃硫酸(H2SO4,北京化工廠,AR).其他所有化學試劑均為分析純,所有溶液均由高純水配制.
1.2 氧化石墨烯與M oS2/Graphene層狀電催化劑的制備
采用改進的Hummers法[14-15]來制備氧化石墨烯(Graphene Oxide,GO).具體合成步驟如下:將1 g石墨鱗片放在一定量的NaCl中一起研磨20min,然后進行抽濾,用大量水反復沖洗石墨粉.將洗后的石墨放在烘箱中70℃干燥30min.干燥的固體石墨粉隨后被轉移到容量瓶中,加入23mL濃硫酸,室溫下攪拌24 h.溶液水浴加熱至40℃,分別加入100 g硝酸鈉(NaNO3)和500 g高錳酸鉀,磁力攪拌30min,同時在反應的過程中隔一段時間加入一定量的水.30min后,將燒瓶從水浴中取出,加入140mL水和10mL 30%的過氧化氫(H2O2),溶液室溫下攪拌5min.將懸浮液進行離心(7500 r/min,15min),得到的沉淀物進行充分洗滌,然后干燥.最終得到黑色的固體,即氧化石墨烯.氧化石墨烯的產率大約為40%.
采用水熱合成的方法來制備MoS2/Graphene層狀電催化劑[11].因為NH2CSNH2在合成過程中做硫源和還原劑,為了研究不同含量的NH2CSNH2對最終形成的MoS2/Graphene的結晶性的影響,本文采用了不同的摩爾濃度的NH2CSNH2來制備MoS2/Graphene復合催化劑,即Na2MoO4·2H2O的濃度為1mmol,NH2CSNH2的濃度分別為 5、6、7和 8 mmol來水熱合成MoS2/Graphene.以 NH2CSNH2用量為 6mmol為例,合成方法如下:取0.013 5 g的氧化石墨烯加入到30mL的二甲基甲酰胺(DMF)中,超聲1 h,然后分別加入 1 mmol的 Na2MoO4·2H2O和6mmol的 NH2CSNH2,混合均勻,放入反應釜中220℃水熱24 h.所得固體分別用無水乙醇和高純水各洗3次,即得到納米級的MoS2/Graphene電催化劑.在水熱過程中NH2CSNH2在高溫下分解放出的H2S將MoO-4和氧化石墨烯還原成MoS2和Graphene.納米MoS2的合成過程同上,只是合成過程中不加入氧化石墨烯.
1.3 材料的表征與性能測試
氧化石墨烯和MoS2/Graphene的形貌使用場發射顯微鏡(FE-SEM,Hitachi S-4800)進行表征.電化學測試均在0.5mol/L的H2SO4中進行,LSV的掃速為1mV/s.電化學工作站為辰華CHI 750C型,采用三電極體系,對電極和參比電極分別為飽和甘汞和大塊Pt片,工作電極為MoS2/Graphene/FTO或 MoS2/FTO.以 MoS2/Graphene為例,工作電極的制備方法為:取10mg的MoS2/Graphene粉末溶于2mL無水乙醇中,超聲1 h,然后旋涂到摻雜氟的SnO2透明導電玻璃(FTO)電極上(每次10μL,旋涂次數為8~16次).
2 結果討論
2.1 氧化石墨烯及M oS2/Graphene電催化劑的結構表征
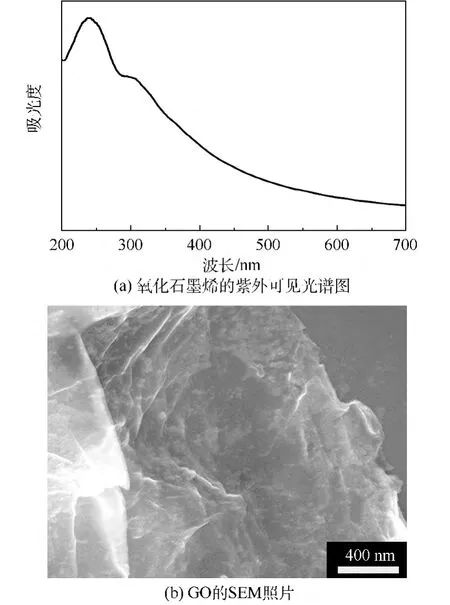
圖1 氧化石墨烯的紫外可見光譜圖與SEM照片Fig.1 UV-vis spectra and SEM photograph of graphene oxide
圖1為氧化石墨烯的紫外可見光譜圖與掃描電鏡(SEM)照片.圖1(a)為用改進的Hummers法制得的氧化石墨烯的紫外可見吸收光譜.氧化石墨烯有2個特征吸收峰,231 nm處的強吸收峰對應于C=C鍵的π→π*躍遷,而300 nm附近的弱吸收峰對應于C=O鍵的n→π*躍遷[16-17].氧化石墨烯的SEM照片如圖1(b)所示.可以看見氧化石墨烯的表面不是很平整,其表面褶皺且其邊緣具有明顯的階梯狀形貌,從圖可清晰觀察到氧化石墨烯特有的二維層狀結構.這確認了本實驗中制備出了氧化石墨烯.
圖2為MoS2/Graphene復合催化劑的SEM照片.MoS2/Graphene電催化劑呈現出二維的火花狀形態,從圖2(b)中也可以看到少量片狀的石墨烯,證明用水熱方法成功地將MoS2和石墨烯合成到了一起.且火花狀的MoS2的邊緣大量的暴露在了外面,這也有利于電催化制氫反應活性的提高[8].

圖2 MoS2/Graphene復合催化劑的SEM照片Fig.2 SEM photographs of MoS2/Graphene composite catalyst
圖3為MoS2/Graphene復合催化劑的高分辨率的透射電鏡(HRTEM)和透射電鏡(TEM)照片.圖3(a)中0.62 nm的晶格條紋間距對應于MoS2的(002)晶面[10],0.34 nm 的晶格條紋間距對應于 Graphene 的(002)晶面[18].從圖 3(b)中可以清晰地看出層狀的MoS2有序緊密地分散在Graphene上,而不是 MoS2雜亂無序地堆疊在Graphene上.這說明水熱合成過程中 MoS2與Graphene之間有著強烈的耦合作用,因而使得MoS2可以選擇性地生長在石墨烯上,從而充分暴露其邊緣的活性位點.
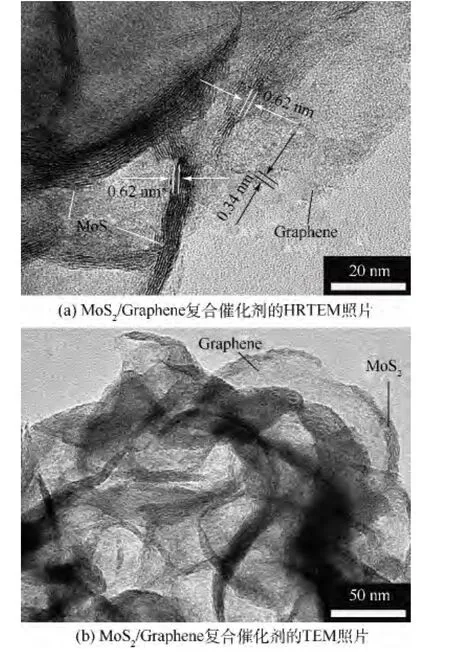
圖3 MoS2/Graphene復合催化劑的HRTEM和TEM照片Fig.3 HRTEM and TEM photographs of MoS2/Graphene composite catalyst,respectively
圖4為用不同摩爾含量的NH2CSNH2做硫源和還原劑,經水熱反應最終形成的 MoS2/Graphene電催化劑的X射線衍射(XRD)圖譜.從中可以看出,不論NH2CSNH2的含量為多少,在2θ =26°處均可觀察到石墨烯的特征峰(002)[19],由此證明了在水熱過程中氧化石墨烯被還原成了石墨烯.由XRD圖譜也可知所制備的MoS2為六方晶系(JCPDS No.65-1951).因此,XRD 圖譜清晰地證明了以 Na2MoO4·2H2O為前驅體,NH2CSNH2為硫源和還原劑,用水熱合成法可以成功地制備出MoS2/Graphene復合電催化劑.對比分別用5、6、7和8mmol的 NH2CSNH2制得樣品的XRD發現,在NH2CSNH2用量為6mmol時,樣品的整體峰型更尖銳,其中在 2θ=14.2°的(002)面的峰強也有明顯的提高.表明在NH2CSNH2用量為 6mmol時,MoS2/Graphene的結晶度最好,且較強的(002)衍射峰表明得到的MoS2沿c軸方向的層狀結構生長良好,這也與HRTEM照片中的結果吻合的很好.因此,在后續表征MoS2/Graphene催化劑的電催化制氫性能的實驗中均用的是NH2CSNH2用量為6mmol時所制備的電催化劑.
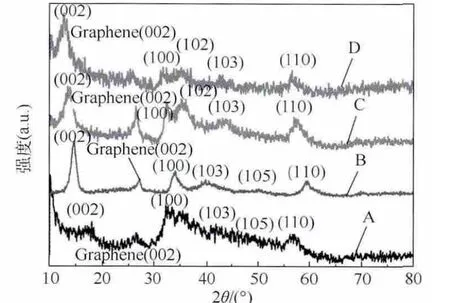
圖4 用不同摩爾含量的NH2 CSNH2做硫源和還原劑,所制備得到的MoS2/Graphene復合催化劑的XRD譜圖Fig.4 XRD image of MoS2/Graphene composite catalysts which were prepared by different molar content of thiourea that served as source of sulfur and reducing agent
2.2 M oS2/Graphene的電催化制氫性能表征
本實驗中將催化劑旋涂到FTO上,采用三電極體系來測定催化劑的電催化制氫性能.圖5為不同電催化劑(體相 MoS2、納米 MoS2、MoS2/Graphene以及Pt)的電催化制氫性能的比較以及旋涂不同層數的MoS2/Graphene催化劑的電催化活性的比較.從圖5(a)中可以看到,體相MoS2幾乎沒有電催化制氫活性[20],而經水熱合成的納米MoS2其電催化制氫活性有了質的提高,其起峰電位提前到了0.15 V左右,在過電位更負時其電流快速地增加,在0.2 V的過電位下其電流密度達到了 -2.1 mA/cm2.當 MoS2與石墨烯復合后,MoS2/Graphene的起峰電位進一步提前到0.1 V,在0.2 V的過電位下其電流密度達到了-3.7mA/cm2,幾乎是純 MoS2的2倍.這是因為在電催化制氫反應中,MoS2的活性位點大多位于MoS2的邊緣上[6,8].體相中 MoS2雜亂無序地堆疊在一起,活性位點少.而經水熱反應合成的納米MoS2具有層狀結構,因此大大地增加了其邊緣的活性位點,所以具有良好的電催化制氫活性.而MoS2與石墨烯復合后,可能是它們之間的良好電化學耦合作用提高了復合催化劑的電催化活性.當然,MoS2/Graphene的電催化制氫性能離Pt還是有一定的差距,但相比貴金屬Pt,MoS2更為便宜,原料來源更為豐富,因此具有良好的發展與應用前景.
圖5(b)為旋涂不同層數的MoS2/Graphene催化劑的電催化活性的比較.可以看到,隨著旋涂層數的增加,其電催化制氫活性先增加后減小.當旋涂層數為12層時,MoS2/Graphene電催化劑具有最佳的活性,其起峰電位為0.085 V,在0.2 V的過電位下其電流密度達到了 -4.5mA/cm2.MoS2/Graphene催化劑層數的增加對電催化制氫活性有兩方面的影響:①MoS2/Graphene催化劑在FTO上的厚度越厚,其表面積會越大,活性位點相應的也就會增多,因此電流密度也會增大;②厚度的增加會造成催化劑和FTO基底之間的導電性變差,電子轉移阻抗變大[21],這會降低催化劑的電催化活性.因此,從催化活性的角度考慮,12層是MoS2/Graphene電催化劑旋涂到FTO上的最佳層數,此時陰極電流密度最大.
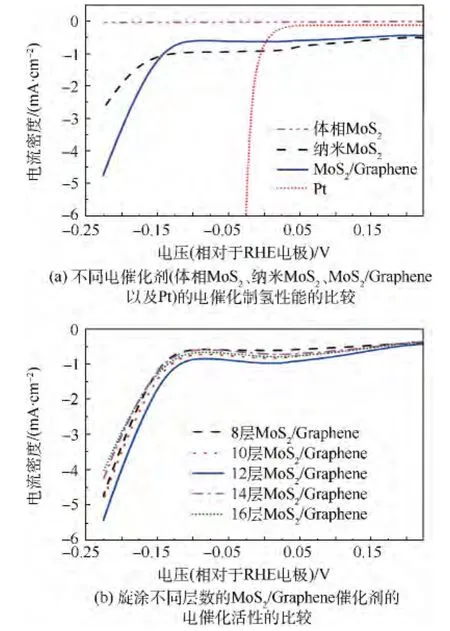
圖5 不同電催化劑(體相MoS2、納米MoS2、MoS2/Graphene以及Pt)以及旋涂不同層數的MoS2/Graphene催化劑的極化曲線Fig.5 Polarization curves obtained with different catalysts(bulk MoS2,nano MoS2,MoS2/Graphene and Pt)and polarization curves obtained with different layered MoS2/Graphene
圖6為對MoS2/Graphene電催化劑的穩定性進行測試.電催化制氫的穩定性是衡量一個催化劑性能好壞最重要的指標之一.為此,本實驗中對MoS2/Graphene復合電催化劑循環不間斷地掃描1000圈.可以看出,第1000圈和最初的第1圈相比,起峰電位幾乎沒有變化,陰極的電流密度的損失也幾乎可以忽略不計.因此,實驗中所制備的MoS2/Graphene復合電催化劑具有非常好的穩定性[22].
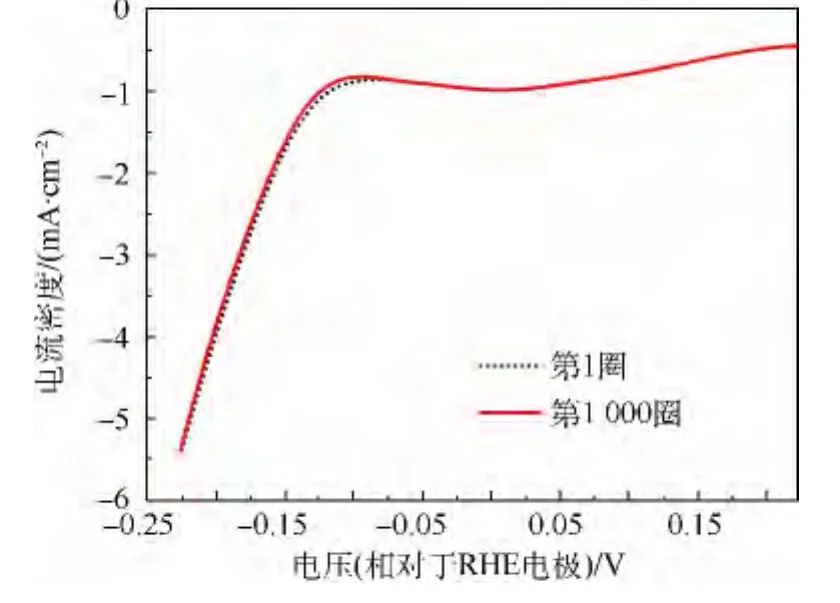
圖6 MoS2/Graphene電催化劑的穩定性測試Fig.6 Durability test for MoS2/Graphene electrocatalyst
2.3 M oS2/Graphene電催化制氫的機理分析
塔菲爾斜率是電催化材料的內在特性,它是由電催化制氫反應的限速步驟所決定.對塔菲爾斜率進行闡釋和說明對理解電催化反應的內在機理非常重要.納米 MoS2、MoS2/Graphene以及 Pt的塔菲爾曲線圖如圖7所示.塔菲爾曲線的公式為

式中:η為過電位;j為電流密度;b為塔菲爾斜率;a為j=1 mA·cm-2時的過電勢(塔菲爾曲線與過電位軸的截矩).
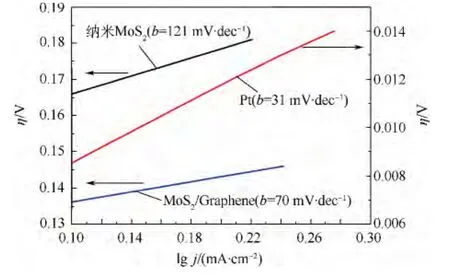
圖7 納米MoS2、MoS2/Graphene以及Pt催化劑的塔菲爾曲線Fig.7 Tafel plots for nano MoS2,MoS2/Graphene and Pt catalysts
因此,由圖 7可以得出納米 MoS2、MoS2/Graphene以及Pt的塔菲爾斜率分別為121、70和31mV·dec-1.
在酸性溶液中,電催化制氫反應有3步可能的反應步驟[23]:
1)質子放電(discharge reaction)步驟:

式中:R為理想氣體常數;T為絕對溫度;α≈0.5為對稱系數;F為法拉第常數;Hads為吸附態氫離子.
2)催化復合(combination reaction)步驟:
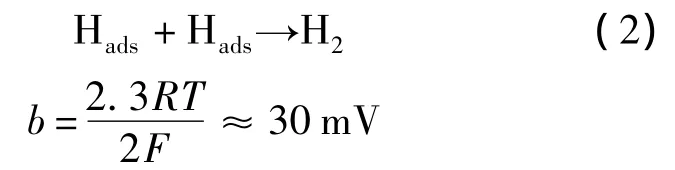
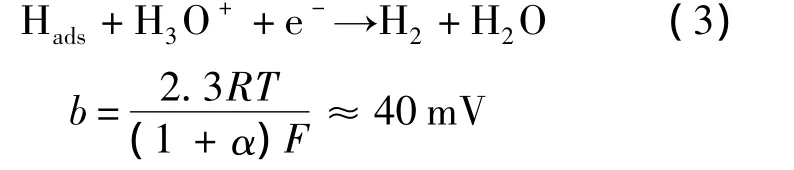
由此可知,電化學制氫反應中,由于Pt的塔菲爾斜率為31mV·dec-1,因此在Pt表面發生的反應是步驟1)和步驟2),即快速的質子放電步驟1)和一個限速步驟即催化復合步驟2).然而,從發現MoS2對電化學制氫有良好的催化效果至今[24],MoS2電催化的反應機理仍未搞清楚.在本實驗中,在納米MoS2表面,由于其塔菲爾斜率為121mV·dec-1,因此可以推測步驟1)可能為其限速步驟[25].MoS2/Graphene 的塔菲爾斜率為70mV·dec-1,文獻[8,26]中也有所報道.由Thomas的研究[27]可知,在電催化反應中較大的塔菲爾斜率(~60mV·dec-1)意味著在化學吸附制氫過程中需要更多的活化能.因此,MoS2/Graphene電催化制氫的可能機理為:一個快速的質子放電步驟1),后續步驟為步驟2)或步驟3),即催化復合步驟2)或電化學脫附步驟3)均有可能為其限速步驟.單純地從塔菲爾斜率上不能判斷為哪一種機理.
圖8為不同電催化劑的電化學阻抗圖譜.圖8(a)中旋涂層數為12層的MoS2/Graphene具有最小的電子轉移阻抗,更小的電子轉移阻抗意味著更快的電子轉移速度[22],因此圖5(b)中旋涂層數為12層的MoS2/Graphene催化劑才會具有最好的電催化制氫活性.且不同旋涂層數電催化劑的電子轉移阻抗變化的趨勢和圖5(b)中催化劑的電催化制氫活性的變化是相一致的.圖8(b)中表明,MoS2/Graphene的電子轉移阻抗Zf(Zf≈150Ω)比納米 MoS2的(Zf≈5 kΩ)要小得多.從圖8(b)的局部放大圖可以看出Graphene
3)電化學脫附(electrochemical desorption reaction)步驟:的電化學阻抗譜包含一個半圓和一個斜直線.低頻區的斜直線可以看作Graphene的純電容行為,而高頻區的半圓則代表其電子轉移阻抗[28-29],約為5Ω.因此石墨烯具有非常低的電子轉移阻抗,所以其能作為良好的導電基體大大加快電子由層狀邊緣的活性位點轉移到溶液中的速度.因而MoS2/Graphene具有比MoS2好得多的電催化制氫活性.

圖8 在過電位 η=0.12V下,0.5mol/L的H2 SO4溶液中,不同旋涂層數的MoS2/Graphene的電化學阻抗圖譜及納米MoS2、MoS2/Graphene和Graphene的電化學阻抗圖譜Fig.8 AC impedance spectroscopy of different layered MoS2/Graphene and nano MoS2、MoS2/Graphene and Graphene catalysts in 0.5mol/L H2 SO4 atη =0.12V
圖9為MoS2和石墨烯經水熱方法合成到一起的機理示意圖.由HRTEM可以看出,MoS2是通過化學耦合和電子耦合選擇性的生長在石墨烯基底上[10].MoS2/Graphene良好的電催化制氫性能正是來源于此.一方面,通過化學耦合作用,生長在基底上的MoS2有相對大的比表面積,這使得層狀MoS2表面富有更多的邊緣,因此大大增加了電催化制氫的活性位點.另一方面,石墨烯作為良好的電子導體,能夠加快電子的轉移速度,使電子更快地由層狀邊緣的活性位點轉移到溶液中,從而提高電催化制氫的活性.
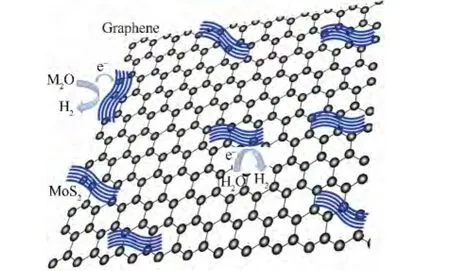
圖9 MoS2/Graphene復合催化劑里的電子轉移機理圖Fig.9 Mechanism schematic diagram ofthe charge transfer in MoS2/Graphene composite catalysts
3 結論
1)采用水熱合成的方法,以Na2MoO4·2H2O為前驅體,NH2CSNH2為硫源和還原劑,合成了層狀的MoS2/Graphene電催化劑.MoS2通過化學耦合選擇性地生長在石墨烯上,使得MoS2邊緣擁有豐富的活性位點,石墨烯作為良好的導電基體也能大大加快電子由層狀邊緣的活性位點轉移到溶液中的速度.
2)MoS2/Graphene表現出了良好的電催化制氫性能,其起峰電位為0.085 V,在0.2 V的過電位下其電流密度達到了-4.5mA/cm2,塔菲爾曲線的斜率約為70mV·dec-1.
3)MoS2/Graphene復合催化劑因其高的電催化制氫效率以及低的成本,很有希望成為Pt族金屬的替代品,具有廣闊的應用前景.
References)
[1] Turner JA.Sustainable hydrogen production[J].Science,2004,305(5686):972-974.
[2] Dresselhaus M,Thomas I.Alternative energy technologies[J].Nature,2001,414(6861):332-337.
[3] Barreto L,Makihira A,Riahi K.The hydrogen economy in the 21st century:A sustainable development scenario[J].International Journal of Hydrogen Energy,2003,28(3):267-284.
[4] le Goff A,Artero V,Jousselme B,et al.From hydrogenases to noblemetal-free catalytic nanomaterials for H2production and uptake[J].Science,2009,326(5958):1384-1387.
[5] Trasatti S.Electrocatalysis of hydrogen evolution:Progress in cathode activation[J].Advances in Electrochemical Science and Engineering,1992,2:1-85.
[6] Laursen A B,Kegn?s S,Dahl S,etal.Molybdenum sulfides-efficient and viable materials for electro-and photoelectrocatalytic hydrogen evolution[J].Energy & Environmental Science,2012,5(2):5577-5591.
[7] Merki D,Hu X.Recent developments of molybdenum and tungsten sulfides as hydrogen evolution catalysts[J].Energy & Environmental Science,2011,4(10):3878-3888.
[8] Jaramillo T F,J?rgensen K P,Bonde J,et al.Identification of active edge sites for electrochemical H2evolution from MoS2nanocatalysts[J].Science,2007,317(5834):100-102.
[9] Hinnemann B,Moses P G,Bonde J,et al.Biomimetic hydrogen evolution:MoS2nanoparticles as catalyst for hydrogen evolution[J].Journal of the American Chemical Society,2005,127(15):5308-5309.
[10] Li Y,Wang H,Xie L,et al.MoS2nanoparticles grown on graphene:An advanced catalyst for hydrogen evolution reaction[J].Journal of the American Chemical Society,2011,133:7296-7299.
[11] Chang K,Chen W.In situ synthesis of MoS2/graphene nanosheet composites with extraordinarily high electrochemical performance for lithium ion batteries[J].Chemical Communications,2011,47(14):4252-4254.
[12] Wang T,Liu L,Zhu Z,et al.Enhanced electrocatalytic activity for hydrogen evolution reaction from self-assembled monodispersed molybdenum sulfide nanoparticles on an Au electrode[J].Energy & Environmental Science,2013,6(2):625-633.
[13] Wang X,Yan Y,Ge X,etal.Facile synthesis of low crystalline-MoS2nanosheet-coated CNTs for enhanced hydrogen evolution reaction[J].Nanoscale,2013,5(17):7768-7771.
[14] Dikin D A,Stankovich S,Zimney E J,et al.Preparation and characterization of graphene oxide paper[J].Nature,2007,448(7152):457-460.
[15] Liang Y,Li Y G,Wang H,et al.Co3O4nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction[J].Nature Materials,2011,10(10):780-786.
[16] Marcano D C,Kosynkin D V,Berlin JM,et al.Improved synthesis of graphene oxide[J].ACS Nano,2010,4(8):4806-4814.
[17] Li D,Muller M B,Gilje S,et al.Processable aqueous dispersions of graphene nanosheets[J].Nat Nanotechnol,2008,3(2):101-105.
[18] Xiang Q,Yu J,Jaroniec M.Synergetic effect of MoS2and graphene as cocatalysts for enhanced photocatalytic H2production activity of TiO2nanoparticles[J].Journal of the American Chemical Society,2012,134(15):6575-6578.
[19] Liu C J,Tai S Y,Chou SW,et al.Facile synthesis of MoS2/graphene nanocomposite with high catalytic activity towardtriiodide reduction in dye-sensitized solar cells[J].Journal of Materials Chemistry,2012,22(39):21057-21064.
[20] Liu Y D,Ren L,Qi X,et al.Preparation,characterization and photoelectrochemical property of ultrathin MoS2nanosheets via hydrothermal intercalation and exfoliation route[J].Journal of Alloys and Compounds,2013,571:37-42.
[21] Guo X,Diao P,Xu D,et al.CuO/Pd composite photocathodes for photoelectrochemical hydrogen evolution reaction[J].International Journal of Hydrogen Energy,2014,39(15):7686-7696.
[22] Liao L,Zhu J,Bian X,et al.MoS2formed on mesoporous graphene as a highly active catalyst for hydrogen evolution[J].Advanced Functional Materials,2013,23(42):5326-5333.
[23] Conway B,Tilak B.Interfacial processes involving electrocatalytic evolution and oxidation of H2,and the role of chemisorbed H[J].Electrochimica Acta,2002,47(22):3571-3594.
[24] Tributsch H,Bennett J.Electrochemistry and photochemistry of MoS2layer crystals.[J].Journal of Electroanalytical Chemistry and Interfacial Electrochemistry,1977,81(1):97-111.
[25] Bonde J,Moses PG,Jaramillo T F,et al.Hydrogen evolution onnano-particulate transition metal sulfides[J].Faraday Discussions,2009,140(1):219-231.
[26] Ji S,Yang Z,Zhang C,et al.Exfoliated MoS2nanosheets as efficient catalysts for electrochemical hydrogen evolution[J].Electrochimica Acta,2013,109:269-275.
[27] Thomas J.Kinetics of electrolytic hydrogen evolution and the adsorption of hydrogen bymetals[J].Journal of Chemical Society,Faraday Transactions,1961,57:1603-1611.
[28] Casero E,Parra-Alfambra A,Petit-Domínguez M,et al.Differentiation between graphene oxide and reduced graphene by electrochemical impedance spectroscopy(EIS)[J].Electrochemistry Communications,2012,20:63-66.
[29] Tian H,Wang L,Qin X,et al.Influence of hydrophilic properties on capacitive behavior of functionalized graphene[J].Ionics,2014,20(8):1055-1061.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
