Apert綜合征1例
2015-03-07 02:49:31張環劉曉紅
疑難病雜志 2015年3期
張環,劉曉紅
?
罕少見病例
Apert綜合征1例
張環,劉曉紅
Apert綜合征;病例報告;X線計算機攝影術
【DOI】 10.3969 /j.issn.1671-6450.2015.03.031
患兒,男,6歲,因雙手、雙足各指(趾)粘連6年入院。患兒出生時家人即發現雙手2~5指及雙足2~5趾粘連。隨著患兒發育,漸發現面部發育異常,頭顱呈扁頭畸形,眼球外凸,眼距寬,下瞼睫毛內翻,致眼球紅腫、溢淚。查體:患兒身材相對于同齡兒童矮小,身高110cm,體質量19kg,智力低下。心臟聽診未聞及病理性雜音,雙肺聽診呼吸音清,腹部未及異常。專科情況:頭顱發育異常,呈扁平狀,前額寬廣,頭顱橫徑16cm,前后徑15cm。雙眼發育畸形,眼球外凸,內眥間距寬約3.5cm,內外眥角不在同一水平,內眥高于外眥約1cm。雙眼平視時,雙上瞼瞼緣覆蓋瞳孔中央水平線,雙側下瞼瞼睫毛內翻,導致眼球紅腫、溢淚。下頜骨發育異常。患兒平靜時,呈張口狀,流涎,上腭正中可見裂跡。頭顱正側位X線片示:頭顱呈舟狀,顱縫早期閉合,腦回壓跡增多、加深,上頜發育不全,下頜突出,出現低位前牙,牙列擁擠(圖1)。雙手2~5指及雙足2~5趾完全粘連,甲融合,無活動度。雙手、足X線片示:手足分節錯亂,雙手指骨短,拇指僅有1節指骨,左手3~4指指骨融合,其余2~5指周圍軟組織相連,右手2~5指骨遠節指骨聚攏,呈融合狀,其周圍軟組織亦相連,雙腕僅有4塊腕骨(圖2)。雙足趾骨粗短,跗骨發育較小,諸趾骨周圍軟組織相連(圖3)。陰莖、陰囊發育正常, 睪丸未觸及。診斷為Apert綜合征。
討 論Apert綜合征較罕見,為一種散發的常染色體顯性遺傳病,又稱尖頭并指(趾)畸形,發病率1/16萬~1/20萬[1],最早在1906年由法國醫生Apert首先報道而得名。父親年齡較大為高危因素,而本例患兒父母并無高齡因素,考慮存在基因突變可能。有報道稱,由于10q25-10q26的FGFR2基因突變而致病,目前發現的突變有FGFR2Ser252Trp或Pro253Arg,顱面部及骨骼畸形主要與FGFR2Ser252Trp有關,而手足并指(趾)畸形主要與FGFR2Ser253Arg有關[2]。Apert綜合征臨床表現為:(1)頭顱發育畸形,短頭,前額寬、高且扁平,所有病例幾乎均出現顱縫早閉。(2)突眼,兩眼分離過遠且眼裂向外下傾斜;(3)塌鼻梁;(4)張口、牙列擁擠、腭弓高且窄,頜凸畸形;(5)手指并連在一起,呈戴連指手套狀,各足趾并連,呈穿短襪狀;(6)智力發育落后。X線檢查特點:塔形舟狀短頭畸形,幾乎所有病例出現早期顱縫閉合,尤其是冠狀縫。眶窩變淺,下頜凸出明顯,上頜發育不全。手、足各骨分節錯亂,多有骨性連接。其他畸形還可見腦畸形,腦積水,關節僵硬及多指(趾)畸形等。本例患兒身材矮小,智力低下,并具有典型精神面容及顱骨改變、雙手、足2~5指(趾)并指(趾)畸形及進行性骨性連接,符合Apert綜合征的臨床診斷。
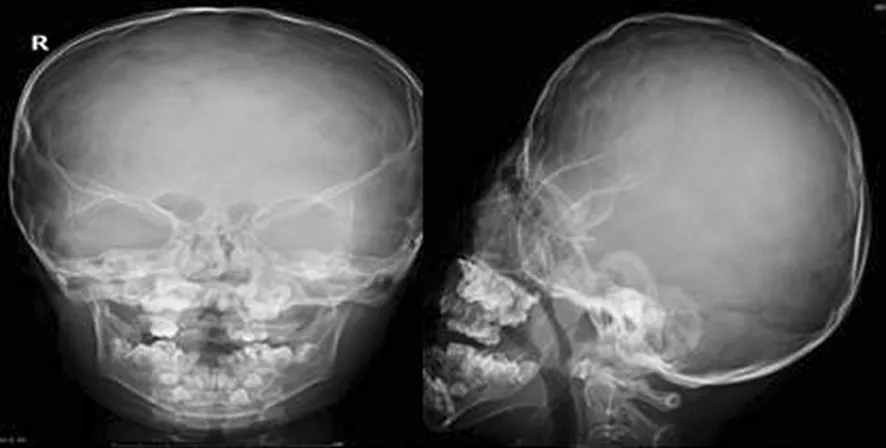
圖1 患兒頭顱X線表現
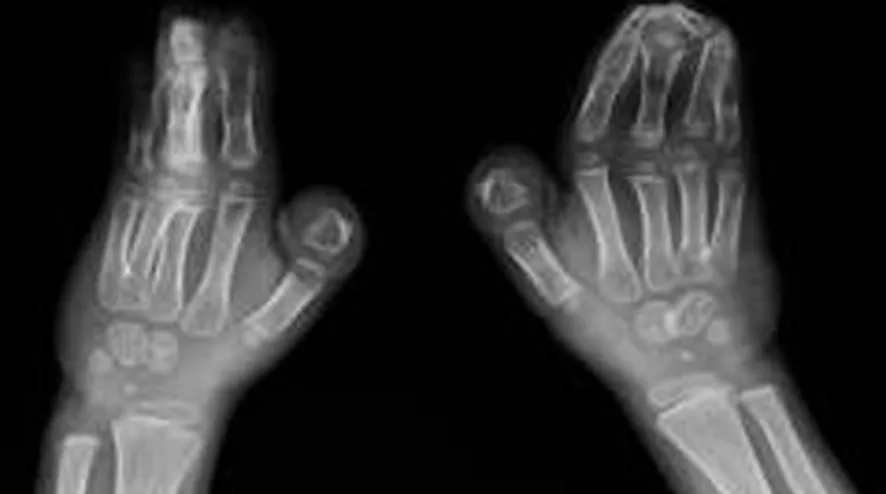
圖2 患兒雙手X線表現
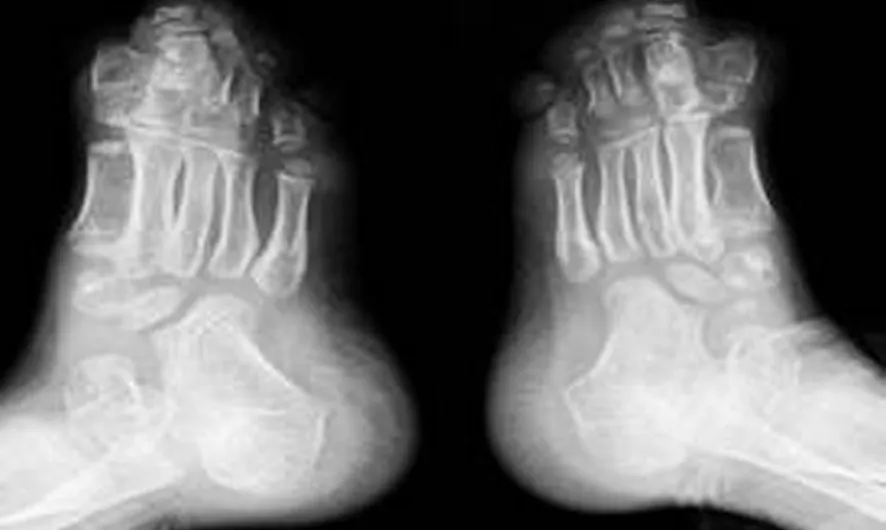
圖3 患兒雙足X線表現
1 葉一蓮,楊永山,簡佩君.Apert綜合癥一例[J].中國新生兒科雜志,2013,28(5):343.
2 李洪華,郝云鵬,杜琳,等.Apert綜合癥一例[J].中國當代兒科雜志,2011,13(7):604.
730050 蘭州軍區蘭州總醫院放射診斷科
2014-11-26)
