基于序列預測蛋白質和RNA的相互作用
2015-02-18 01:30:00李享云
大連交通大學學報 2015年4期
關鍵詞:特征
汪 穎,李享云
(大連交通大學 理學院,遼寧 大連 116028)*
0 引言
蛋白質與RNA的相互作用在許多生理過程中起著重要的作用,RNA參與很多基本的細胞生理過程,如攜帶來自DNA的遺傳信息,參與形成核糖體、拼接體、端粒酶等許多核酸蛋白顆粒的結構,有些RNA還具有酶活性等,幾乎所有的RNA生物功能的發揮都需要蛋白質的共同作用.蛋白質和RNA相互作用的研究為最終探明RNA和蛋白質相互作用的分子機制,從本質上認識相關細胞生理過程起著不可忽視的重要作用.采用實驗的方法預測蛋白質和RNA的相互作用有很大的局限性,或因實驗步驟過多,既耗時又費力,也增加了實驗結果的不穩定性.因此,利用計算的方法預測蛋白質和RNA相互作用成為當前的一大趨勢.
近年來,由于RNA本身的復雜性導致蛋白質和RNA相互作用的研究一直處于滯后狀態.但是隨著實驗獲取的RNA數據以及蛋白質和RNA復合物數據的增加,蛋白質和RNA相互作用的預測方法研究成為目前非常緊迫的一項重要課題.2011 年,Pancaldi和Bahler[1]首次提出了一種預測蛋白質和RNA相互作用的方法,選取100多種顯著性較高的特征(包括Gene Ontology條款,基因和蛋白質的物理性質,mRNA性質,蛋白質的二級結構以及基因的相互作用genetic interactions等)構建特征向量.然而,由于該文中用到的特征種類較多,有些特征不易獲取,所以這種方法具有一定的局限性.同年,Bellucci等人[2]提出一種新的預測蛋白質-RNA相互作用的方法catRAPID,考慮存在于氨基酸鏈和核苷酸鏈中的幾乎所有關聯,從中選取了傾向性較高的二級結構、氫鍵和范德華這三種性質,并基于此計算每個RNA和蛋白質對的相互作用傾向性,用于預測蛋白質和NRA的相互作用.以上兩種方法均考慮了蛋白質和RNA多種性質特征.對于現有的蛋白質-RNA數據而言,都有著一定的局限性.于是在2011年和2013年,文獻[3-4]主要基于蛋白質和RNA序列信息,即氨基酸和核苷酸的成分特征,構建機器學習模型.在研究[4]中,基于蛋白質序列中氨基酸組成成分以及RNA序列中核苷酸組成成分,通過特征選取的方法提取有效特征構建向量,從而構建預測模型.通過對多組數據的預測,證實了特征選取方法以及預測模型的有效性.但是,特征選取方法也存在一個弊端,即被選取的特征在某種程度上依賴于樣本數據.
本文基于蛋白質和RNA序列,提出了一種新的預測蛋白質-RNA相互作用的方法.本文只考慮了氨基酸三聯體和核苷酸的組成成分,利用其成分比率以及氨基酸三聯體-核苷酸相互作用傾向性構建了一種新的用于衡量蛋白質和RNA序列對個體的三聯體-核苷酸傾向性度量,并利用該傾向性以及氨基酸三聯體和核苷酸的成分特征構建支持向量機(support vector machine,SVM)模型,預測其相互作用.
1 預測模型和算法
1.1 氨基酸三聯體-核苷酸的相互作用傾向性
氨基酸三聯體[5]指的是三個連續的氨基酸構成的一個整體.蛋白質序列中共有20種氨基酸,則三聯體的總個數為20×20×20=8 000個;RNA序列中有4種核苷酸,因而共有8 000×4=32 000個氨基酸三聯體-核苷酸組合.在文獻[6]中,針對一組來自于蛋白質數據庫(PDB)的3149個具有相互作用的蛋白質-RNA對,利用文獻[6]中氨基酸三聯體-核苷酸的相互作用傾向性度量,即文獻[6]中式(1),計算得到了氨基酸三聯體-核苷酸的相互作用傾向性值,見附表1.附表1中出示了32 000個三聯體-核苷酸組合的傾向性值,這些值是針對來自PDB的3149個蛋白質-RNA序列對這個整體數據集而言,氨基酸三聯體-核苷酸的相互作用傾向性,在這里稱之為整體三聯體-核苷酸傾向性.
蛋白質和RNA是否相互作用主要取決于氨基酸和核苷酸位點的結合上.因此本文試圖利用氨基酸三聯體-核苷酸的傾向性這一性質構建特征向量.為了更好地度量每一對蛋白質-RNA序列個體對中三聯體-核苷酸的相互作用傾向性,我們重新定義了一個權重傾向性度量,也可稱為個體三聯體-核苷酸傾向性度量,如下:
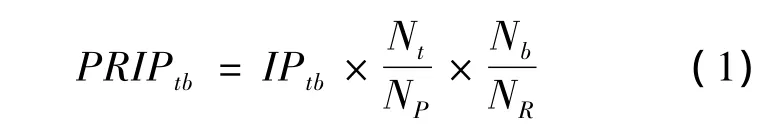
其中,P表示蛋白質序列;R表示RNA序列;t表示蛋白質序列P中的氨基酸三聯體;b表示RNA序列R中的核苷酸;Nt,Nb分別表示蛋白質序列P中氨基酸三聯體t的數量和RNA序列中核苷酸b的數量;NP,NR表示蛋白質序列P中所有氨基酸三聯體的數量和RNA序列中所有核苷酸的數量;IPtb表示由文獻[6]中整體三聯體-核苷酸傾向性度量公式(1)計算得到的三聯體t和核苷酸b的相互作用傾向性值,它表示的是三聯體t和核苷酸b的整體傾向性,而本文中式(1)計算的PRIPtb值表示的是一對蛋白質-RNA序列個體中三聯體t和核苷酸b的相互作用傾向性.以下均用三聯體-核苷酸的整體傾向性表示來自于文獻[6]中的度量公式(1)計算得到的傾向性,即IPtb;用三聯體-核苷酸的個體傾向性表示由本文中的權重傾向性度量公式(1)計算得到的傾向性,即PRIPtb.
1.2 構建特征向量
為了預測一對蛋白質-RNA序列是否相互作用,利用氨基酸三聯體-核苷酸的個體傾向性編譯特征向量.首先,根據極性和側鏈容積等性質,把20種氨基酸分成7類[5],依次是:{A,G,V},{I,L,F,P},{Y,M,T,S},{H,N,Q,W},{R,K},{D,E},{C}.在文獻[5]中,作者利用氨基酸三聯體有效地預測蛋白質-蛋白質相互作用.本文中也同樣使用三聯體特征.20種氨基酸被分成7類,此時三聯體共有7×7×7=343類,依次可以計算出三聯體-核苷酸的組合個數為343×4=1372.給定一對蛋白質-RNA序列,構造如下特征向量:
1.2.1 個體氨基酸三聯體-核苷酸傾向性
第一,利用整體傾向性度量公式[6]分別計算出所有32 000個三聯體-核苷酸組合的相互作用傾向性值IPtb;
第二,基于32000個整體傾向性IPtb,計算每類三聯體-核苷酸傾向性的均值,用來表示這類三聯體-核苷酸的傾向性值,共有343×4=1372個傾向性;
第三,針對每一對蛋白質-RNA序列,利用權重傾向性度量公式(1)計算這對序列中每類三聯體-核苷酸的個體傾向性,并以此作為特征向量.此時式(1)中的IPtb表示的是由第二步計算得到的每類三聯體-核苷酸傾向性均值,Nt表示的是蛋白質序列中每類三聯體的數量;
第四,考慮到組合特征的冗余性,從中選擇具有較高傾向性的三聯體-核苷酸組合,并以這些三聯體-核苷酸組合為基礎建立特征向量.
1.2.2 氨基酸三聯體和核苷酸成分特征
第一,對于一個蛋白質序列,計算343類三聯體的成分比率;
第二,對于一個RNA序列,計算4種核苷酸的成分比率.
1.3 樣本數據
為了證明預測的有效性,本文主要針對兩組不同種類的數據集進行預測:一組是來自NPInter數據庫(http://www.bioinfo.org.cn/NPInter)的367對長鏈非編碼RNA(簡稱ncRNA)和蛋白質相互作用的數據集PRI367,見表1;另一組是來自PRIDB[7]的非冗余數據集PRI369[2],見 表2.PRIDB是一個從PDB[8]里提取的一個綜合的蛋白質和RNA復合物的數據庫.
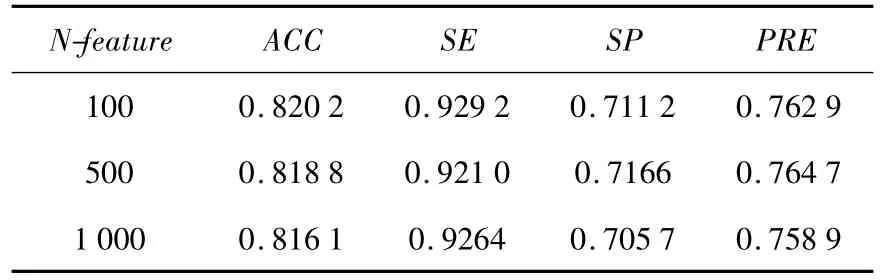
表1 RPI367計算結果
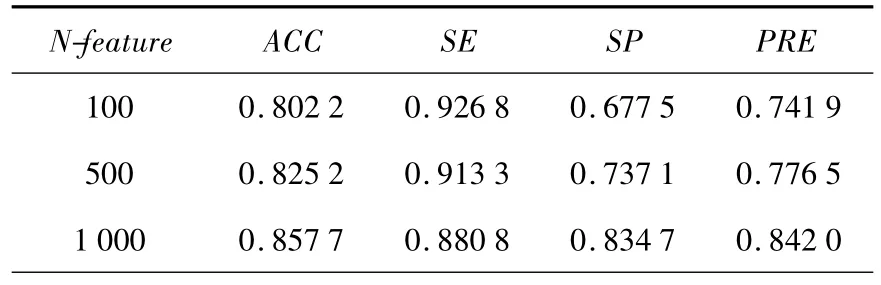
表2 RPI369計算結果
負樣本數據的選取我們采納大多數文獻中的方法,即隨機選取與正樣本數據相同數量的蛋白質和RNA序列對作為負樣本數據集,但是前提是排除那些已知有相互作用的蛋白質-RNA對.
1.4 預測模型和算法
支持向量機(SVM)是Vapnik等人提出的一類新型機器學習方法,是基于統計學習理論、根據結構風險最小化原理而推導出來的.由于SVM出色的學習能力,普遍應用于生物信息學研究中,很多生物信息學中的分類問題都是利用SVM進行分類的.本文也采用SVM對蛋白質-RNA是否有相互作用進行分類預測.
這里簡單地介紹一下支持向量分類機的模型:
對于一個給定的訓練集
T={(x1,c1),(x2,c2),…,(xl,cl)}
其中,xi=(xi1,xi2,…,xin)T∈Rn是輸入(input),表示第i個輸入樣本的n個特征;ci∈{-1,+1}是輸出(output),表示第i個樣本所屬的分類.引入從輸入空間到Hilbert空間的映射φ:Rn→H.支持向量機就是為了尋找一個Hilbert空間的超平面(ω·φ(x))+b=0,使得在最大間隔的基礎上將樣本盡可能的分開.通過使用核函數替代樣本在Hilbert空間中的內積,來判別樣本所屬類別.考慮到RBF核函數優于其他核函數,本文使用RBF核函數.
本文利用公開軟件LibSVM(version 3.18)訓練SVM中的C-SVC,其性能依賴于參數的選擇,所需選擇的參數為:C和gamma.其中C是懲罰參數,是對錯分點的懲罰;gamma是RBF核函數中的參數,它決定向量機的推廣能力.
2 計算結果及討論
在計算中,參數 C=200和gamma=0.1.使用10折交叉驗證程序評價我們的預測算法,預測結果的有效性主要考慮了以下幾個指標:
ACC=(TP+TN)/(TP+FP+TN+FN)
SE=TP/(TP+FN)
SP=TN/(TN+FP)
PRE=TP/(TP+FP)
其中,TP表示真的正樣本(true positives);TN表示真的負樣本(true negatives);FP表示假的正樣本(false positives)和FN表示假的負樣本(false negatives);ACC(正確率),SE(靈敏度),SP(特指度)和PRE(精度).
針對兩組數據集PRI367和PRI369,分別取100,500,1000個三聯體-核苷酸組合特征建立特征向量進行計算,結果見表1和表2.其中N-feature表示所選取的三聯體-核苷酸傾向性的個數.由計算結果可以看出,隨著所選特征的增加,RPI369的正確率有一定的提高,而RPI367的正確率反而降低了.當特征個數增加到1 000時,正確率沒有太大的變化.而在目前僅僅基于序列預測蛋白質 -RNA相互作用的工作[3]中,對RPI369數據集使用兩種分類方法(RF和SVM)實施10折交叉驗證,正確率分別為76.2%和72.8%.在以前的研究工作中,基于 Na?ve Bayesian的分類方法對RPI367和RPI369進行10折交叉驗證,正確率僅僅達到77.6%和75.0%.通過比較,可以看出本文的計算結果更好一些.
氨基酸三聯體和核苷酸的相互作用傾向性被用于預測RNA結合位點得到了很好的預測結果,于是我們試圖把它運用到蛋白質-RNA相互作用的預測中.考慮到每對蛋白質-RNA序列中三聯體-核苷酸的傾向性的差別,重新定義了一個權重傾向性度量,然后利用此度量計算每類三聯體-核苷酸的傾向性.計算結果證實了本文所選特征的有效性,同時也說明了三聯體-核苷酸的相互作用傾向性在蛋白質-RNA相互作用預測中起著不可忽視的重要作用.
[1]PANCALDI V,BAHLER J.In silico characterization and prediction of global protein RNA interactions in yeast[J].Nucleic Acids Res.,2011,39:5826-5836.
[2]BELLUCCI M,AGOSTINI F,MASIN M,et al.Predicting protein associations with long noncoding RNAs[J].Nat.Methods,2011(8):444-445.
[3]MUPPIRALA U K,HONAVAR V G,DOBBS D.Predicting RNA-protein interactions using only sequence information[J].BMC Bioinformatics,2011,12:489.
[4]WANG Y,CHEN X W,LIU Z P,et al.De novo prediction of RNA-protein interactions from sequence information[J].Mol.BioSyst.,2013(9):133-142.
[5]SHEN J,ZHANG J,LUO X,et al.Predicting proteinprotein interactions based only on sequences information[J].Proc.Natl.Acad.Sci.U.S.A.,2007,104:4337-4341.
[6]CHOI S,HAN K.Prediction of RNA-binding amino acids from protein and RNA sequences.BMC Bioinformatics[J].2011,12(Suppl 13):7.
[7]LEWIS BA,WALIA R R ,TERRIBILINI M ,et al.PRIDB:a Protein-RNA Interface Database[J].Nucleic Acids Res.,2011,39:80-82.
[8]BERMAN H M ,WESTBROOK J,FENG Z,et al.The Protein Data Bank[J].Nucleic Acids Res.,2000,28:235-242.
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
