1,3-二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷的合成
2014-12-27 01:18:16劉秀英潘金鵬
武漢紡織大學學報 2014年6期
方 進,劉秀英,潘金鵬
1,3-二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷的合成
方 進,劉秀英*,潘金鵬
(武漢紡織大學 化學與化工學院,湖北 武漢 430200)
采用1,1,3,3-四甲基二硅氧烷和烯丙基縮水甘油醚為原料,以氯鉑酸為催化劑,以四氫呋喃為溶劑,在溫和條件下經硅氫加成合成了1,3-二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷。產物經紅外光譜、核磁共振氫譜和質譜表征。考查了催化劑用量、反應溫度、溶劑、原料摩爾比和反應時間等因素對反應轉化率的影響。當1,1,3,3-四甲基二硅氧烷和烯丙基縮水甘油醚的摩爾比為1:3,催化劑用量為1,1,3,3-四甲基二硅氧烷質量的0.016%,在0-10℃下于四氫呋喃溶劑中攪拌反應12 h,產物摩爾轉化率可達89.5%。
1,3-二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷;硅氫加成;1,1,3,3-四甲基二硅氧烷;烯丙基縮水甘油醚
環氧烴基二硅氧烷是一類端基含環氧基反應性官能團的有機硅二聚體,是制備有機硅和聚氨酯、聚碳酸酯、聚酯及環氧樹脂等的嵌段或接枝共聚物的原料之一[1-8]。由這類二聚體形成的共聚物在分子結構中含有Si-O-Si鏈,因而在成膜性能、抗水性能、介電性能以及耐熱耐寒等方面有較好的特性[2-9]。
合成這類端基為反應性官能團的二硅氧烷的方法通常有硅氫加成法、鈉縮合法及取代反應法等,例如,柴子斌、Speier、Yarosh等通過硅氫加成法合成了雙羥丙基和雙乙烯基等封端的1, 1, 3, 3-四甲基二硅氧烷[1, 10-13];Watanabe、任海云和方少明等則分別用鈉縮合法合成了1, 3-雙苯基、1, 3-雙乙烯基和1, 3-雙羥丙基等四甲基二硅氧烷[14-17];姜紅芹、游革新等用取代反應法合成了端雙羥丙基、端雙羥丁基等四甲基二硅氧烷[8, 18];彭萬華等也采用取代法合成了烯丙基二硅氧烷[9]。
本文擬通過硅氫加成反應合成雙環氧烴基封端的二硅氧烷,即1,3二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷,文獻雖曾報道了該化合物的合成,但需在甲苯體系中高溫條件下經硅氫加成得到,且產率不高[2]。本文探索在溫和條件下較高產率地合成該化合物的方法,擬采用1,1,3,3-四甲基二硅氧烷(TMDS)和烯丙基縮水甘油醚為原料,反應式如下:

1 實驗部分
1.1 原料與儀器
TMDS:工業純,質量分數≥98.0%;烯丙基縮水甘油醚:工業純,質量分數≥99.5%;氯鉑酸(H2PtCl6·6H2O):分析純;其他試劑為化學純。
NICOLET 170SX FT-IR紅外光譜儀(美國Nicolet公司);ADVANCE III 400 MHz 核磁共振儀(瑞士Bruker公司)。TRACEMS2000色譜-質譜聯用儀(美國Finnigan公司)。GC9790型氣相色譜儀(海溫嶺儀器廠)。
1.2 實驗方法
在裝有回流冷凝管、滴液漏斗和溫度計的三口燒瓶中,加入四氫呋喃(THF)溶劑44 mL、氯鉑酸5.5 mg和烯丙基縮水甘油醚88.9 mL (0.75 mol),在冰水浴中磁力攪拌2-3 min使氯鉑酸溶解且使混合均勻。同時,為防止反應過程中溫度急劇上升,將44.2 mL TMDS (0.25 mol)用44 mL THF稀釋,并用滴液漏斗于1 h內滴加到反應體系中。在此期間控制反應體系溫度在0-10℃,并保持攪拌。滴加完后繼續在0-10℃攪拌反應12 h。反應結束后,常壓蒸餾回收THF和TMDS,再減壓蒸餾得到1,3-二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷。產物經氣相色譜分析,摩爾轉化率為89.5%。1H NMR(CDCl3, 400MHz),δ: 0.021 (s, 12H, -CH3(a)), 0.454-0.497 (t, 4H, -CH2-(b)), 1.531-1.609 (m, 4H, -CH2-(c)),2.571-2.590 (m, 2H, -CH2(g1/g2)),2.758-2.781 (m, 2H, -CH2(g1/g2)),3.101-3.140 (m, 2H, -CH-(f)),3.335-3.378(q, 2H, -CH2-(e1/e2)),3.384-3.412(q, 2H, -CH2-(d1/d2)),3.425-3.452(q, 2H, -CH2-(d1/d2)),3.659-3.696 (q, 2H, -CH2(e1/e2))。MS(): 363.0 ( M+1)。IR,cm-1:3053、1253 (-CH2-),2959、2927 (-CH3),1105 (C-O-C),1049 (Si-O-Si),836、794 (Si-CH3)。
2 結果與討論
2.1 催化劑用量的影響
以THF為反應溶劑,當原料TMDS與烯丙基縮水甘油醚的摩爾比為1:2.3,反應溫度為0-10 ℃,反應時間為2 h時,改變氯鉑酸用量,其對產物轉化率的影響如圖1。當催化劑用量為TMDS質量的0.008%時,轉化率小于30%;增加催化劑用量時,產物轉化率大幅提高,當用量為0.016%時,轉化率達到81.5%;繼續增加用量時,轉化率變化平緩,且總體上略降。因此選擇催化劑的用量為TMDS質量的0.016%。
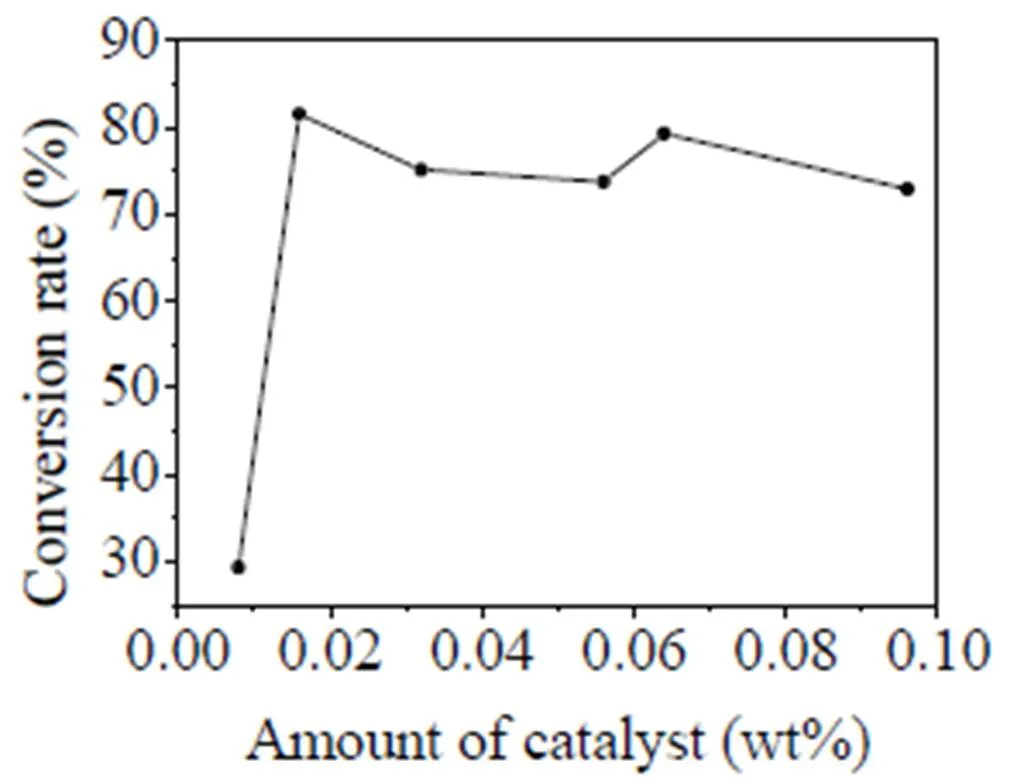
圖1 催化劑用量對轉化率的影響
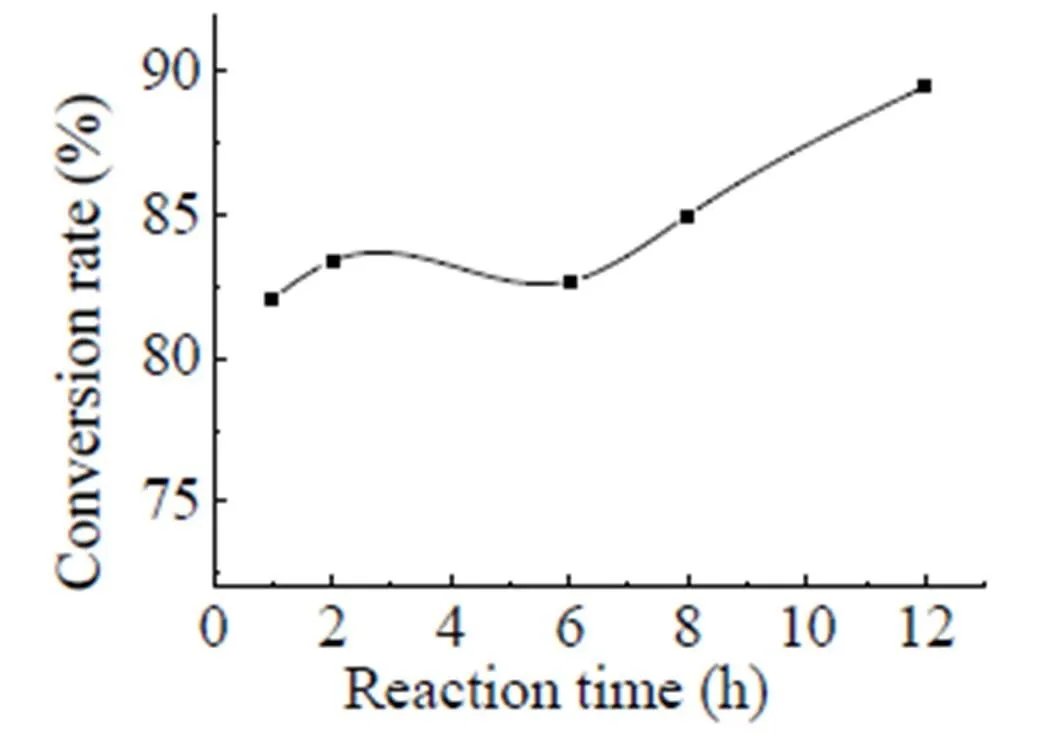
圖2 反應時間對轉化率的影響
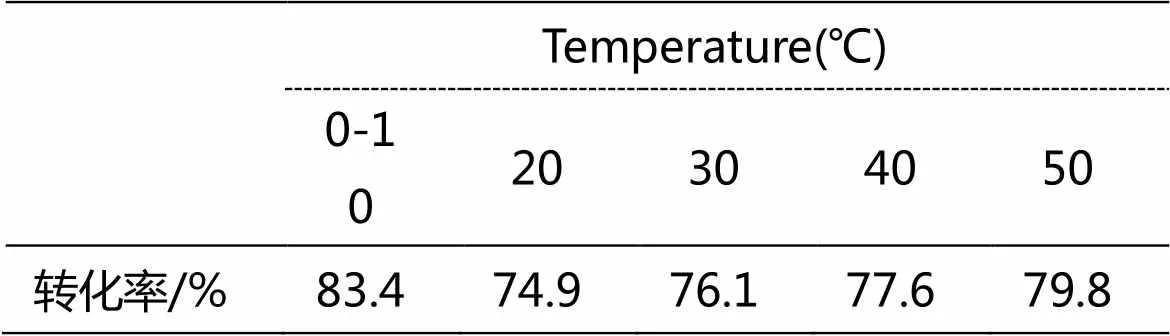
表1 反應溫度對轉化率的影響
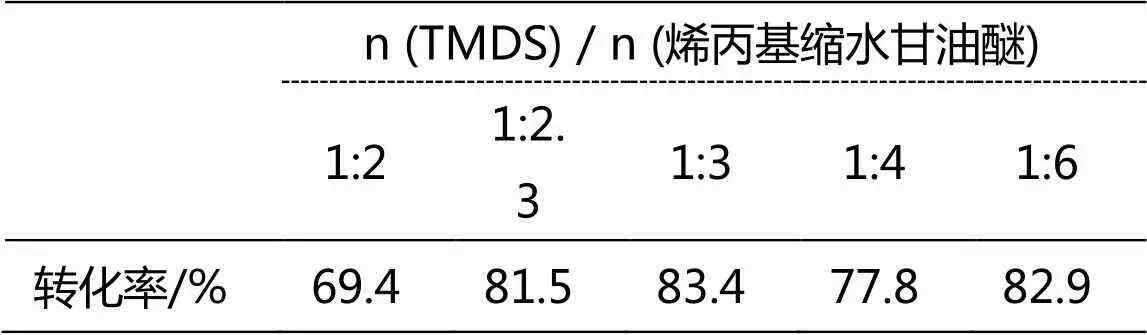
表2 原料摩爾比對收率的影響
2.2 反應溫度的影響
本反應為放熱反應,若不采用溶劑,則在滴加原料的過程中,體系溫度急劇上升至100℃以上。為控制反應的進行,采用低沸點的THF為反應溶劑;同時將原料TMDS用溶劑稀釋后再采用滴加的方式加入。
固定催化劑的用量為TMDS質量的0.016%,TMDS與烯丙基縮水甘油醚的摩爾比為1:3,反應時間為2 h,THF為溶劑,考查了反應溫度對產物轉化率的影響,結果如表1,當反應在低溫0-10℃下進行時可達到較高的轉化率。與已有文獻需在110℃的高溫等條件下制備相比,本方法具有反應條件溫和、能耗低、無需氮氣保護和產物轉化率高等特點[2]。
2.3 反應物摩爾比的影響
在最佳催化劑用量和反應溫度下,固定反應時間為2 h,以THF為溶劑,考查了TMDS與烯丙基縮水甘油醚用量對產物轉化率的影響,如表2,當兩者摩爾比為1:3,即烯丙基縮水甘油醚過量50%時,轉化率最高,可達83.4%。
2.4 反應時間的影響
以THF為溶劑,采用最佳催化劑用量、原料用量摩爾比和反應溫度時,反應時間對產物轉化率的影響如圖2,當反應時間為1-6 h時,轉化率變化不大;繼續延長反應時間,轉化率大幅增大;反應時間為12 h時,轉化率可達89.5%。
3 結論
以TMDS與烯丙基縮水甘油醚為原料,分別采用氯鉑酸和THF為催化劑和反應溶劑,合成了1,3-二[3-(環氧乙基甲氧基)丙基]-1,1,3,3-四甲基二硅氧烷。當TMDS與烯丙基縮水甘油醚摩爾比為1:3,催化劑用量為TMDS質量的0.016%時,在0-10℃下反應12 h,產物摩爾轉化率可達89.5%。該合成方法具有反應條件溫和、能耗低和轉化率較高的優點。
[1] 柴子斌.反應性官能基硅氧烷的合成與表征[D].杭州:浙江大學, 2011.1-68.
[2] Zhu X L,Zhang M, Zhang Q S,et al. Synthesis and characterization of bis(methoxyl hydroxyl)-functionalized disiloxane and polysiloxane oligomers derivatives therefrom[J].European Polymer Journal, 2005, 41:1993-2001.
[3] 李擁有,盤毅,謝凱.端環氧基聚二甲基硅氧烷的合成及表征[J].精細化工中間體,2004,34(1):23-25.
[4] 黃良仙,安秋鳳,楊剛,等.環氧改性硅油的制備與表征及應用進展[J].日用化學工業,2007,37(3):189-211.
[5] 李曉茹,叢麗曉,張圣有,等.聚硅氧烷改性環氧樹脂的研究進展[J].有機硅材料,2005,19(5):33-36.
[6] 張亞光,李會錄,樊淑蘭,等.聚硅氧烷改性環氧樹脂的研究進展[J].現代塑料加工應用,2012,1:61-63.
[7] 鄭欽健,李航昱,陳少鵬,等.含氨基聚硅氧烷改性環氧樹脂[J].廈門大學學報(自然科學版),2005,44(3):399-402.
[8] 姜紅芹,張墩明,蔣錫群,等.1,3-雙(3-羥丙基)-1,1,3,3-四甲基二硅氧烷的制備[J].精細化工,2004,21(3):232-234, 240.
[9] 彭萬華,趙永鎮.烯丙基二硅氧烷的合成及其自由基聚合反應[J].化學通報,1998,7:40-42.
[10]柴子斌,吳清洲,陳關喜,等.羥基保護-硅氫加成合成1,3-雙(γ-羥丙基)-1,1,3,3-四甲基二硅氧烷[J].精細化工,2010,27(9):86-89.
[11]Speier J L, Webster J A, Barnes G H.The addition of silicon hydrides to olefinic double bonds.Part Ⅲ the use of group Ⅷ metal catalysts[J]. Journal of the American Chemical Society,1957,(04):974-979.
[12]Yarosh O G,Zhilitskaya L V,Yarosh N K,et al.3-Bis[2-(ethynyldimethylsilyl)vinyl]tetramethyldisiloxane and macro- cyclic polyunsaturated siloxanes on its basis[J].Russian Journal of General Chemistry, 2005, 75(8):1230-1233.
[13]Tadashi O,Mikami R.Method for the preparation of carbinol group-containing organopolysiloxane.US:5395955A[P], 1995-03-07.
[14]Watanabe H. Phenylazotrioganosilanes as silyated phenyldia- zenes: a convenient precursor for phenldiazene [J]. J Organo-metallic Chemistry,1980,195:363.
[15]姜亞, 吳清洲, 陳關喜, 等.1,3-二苯基-1,1,3,3-四(二甲基硅氧基)二硅氧烷的合成與表征[J].有機硅材料.2010,24(5):263-266.
[16]任海云,蘭支利,尹篤林.1,1,3,3-四甲基-1,3-二乙烯基二硅氧烷的合成[J].化工之友,2006,8:54-56.
[17]方少明,侯守君,程海軍,等.合成l,3-二(3- 羥丙基)四甲基二硅氧烷的新方法[J].合成化學,1996,4(3):265-266.
[18]游革新, 趙耀明,劉海敏,等.二-(4-羥基丁基)四甲基二硅氧烷的合成研究[J],合成材料老化與應用,2003,32(3):6-8,53.
Preparation of 1,3-bis-[(3-oxiranylmethoxy-propyl)]-1,1,3,3-tetramethyl-disiloxane
FANG Jin, LIU Xiu-ying, PAN Jin-peng
( School of Chemistry and Chemical Engineering, Wuhan Textile University, Wuhan Hubei 430200, China)
1,3-bis-(3-oxiranylmethoxy-propyl)-1,1,3,3-tetramethyl-disiloxane was synthesized in mild condition via hydrosilylation reaction between 1,1,3,3-tetramethyl-disiloxane and allyl glycidyl ether. Tetrahydrofuran was used as solvent and chloroplatinic acid as catalyst. The products was characterized by IR, 1H NMR and the mass spectrometry. The effects of the reaction conditions including the catalyst dosage, the reaction temperature, the solvent, the molar ratio of reactants and the reaction time on conversion rates were investigated. The molar conversion rate could reach 89.5% when the reaction conditions were as followings: the molar ratio of 1,1,3,3-tetramethyl-disiloxan to allyl glycidyl ether was 1:3, the amount of the catalyst was 0.016 % based on the weight of 1,1,3,3-tetramethyl-disiloxan, and the reaction was stirred at 0-10℃ for 12 h in tetrahydrofuran.
1,3-bis-[(3-oxiranylmethoxy-propyl)]-1,1,3,3-tetramethyl-disiloxane; Hydrosilylation; 1,1,3,3-tetramethyl-disiloxan; Allyl Glycidyl Ether
劉秀英(1971-),女,副教授,博士,研究方向: 有機合成.
國家自然科學基金項目(51103110).
O 626.15
A
2095-414X(2014)06-0061-03
