55%Al-Zn合金熔體結構的從頭算研究
2014-11-16 07:50:12邢文國孟憲興馮維春張長橋
航空材料學報 2014年2期
關鍵詞:結構
邢文國, 孟憲興, 馮維春, 張長橋
(1. 山東省生物化學工程重點實驗室,濟南250014;2. 山東大學 化學與化工學院,濟南250061)
鋼鐵表面熱鍍耐蝕金屬鍍層,是其防止大氣腐蝕的主要方法之一[1]。鋼鐵表面熱鍍耐蝕金屬物理機制的研究成為實驗和理論工作者共同關注的問題。較早發展和應用起來的是熱鍍純鋅鍍層,但其耐蝕性能有限。熱鍍鋁鍍層的耐蝕效果明顯優于熱鍍鋅,但由于鋁的熔點較高,化學活性大,鋁液與鋼鐵表面浸潤性差,而使得鍍層質量和結構控制不夠穩定。最近幾年發展起來的熱鍍鋁鋅合金,綜合了純鋅、純鋁鍍層各自的特點,具有良好的耐腐蝕性和較好的陰極保護性能,鍍層質量易于控制,鍍層結構穩定。目前,研究和發展較快的有5%Al-Zn 的Galfan 和55%Al-Zn 的Galva1um 合金鍍層[2~9]。尤其是后者,在工業大氣中其耐蝕性比純鋅鍍層優良得多,與純鋁鍍層相近。
實驗發現5%Al-Zn 和55%Al-Zn 合金鍍層可以產生較好的耐蝕性,但是鋁含量對Al-Zn 合金耐蝕性能的影響不遵從線性規律。目前對這一問題的物理機制并不清楚,其難點在于該合金的高溫熔體結構以及冷卻過程直接影響最終耐蝕效果,因此深入了解該合金熔體的結構及電子結構,以此為基礎進一步研究冷凝過程中其結構的變化規律,對于材料的成分設計和尋找最佳工藝條件以獲得更好的耐蝕性能有著非常重要的實際意義。實驗上可以使用X 射線衍射或中子散射等技術得到合金熔體的結構參數,但很難給出熔體局域結構細節的描述。另外,由于合金熔體溫度較高,實驗難度比較大。
第一性原理分子動力學模擬方法可以給出熔體結構的描述。相對于經典的分子動力學,該方法的優點在于不需要任何調整參數,并對電子結構進行優化,原子間的相互作用對化學環境的依賴性不強,可以給出原子成鍵的變化對熔體局域結構的影響,現已成為了解熔體微觀結構信息的主要手段之一,并取得了一系列進展[10~12]。
本工作采用第一性原理分子動力學模擬方法對高溫55%Al-Zn 合金熔體進行了研究。給出了熔體的結構和動力學性質,為進一步研究其冷卻過程變化規律,從而更有效地確定加工工藝,提供理論基礎。
1 計算方法
計算使用VASP 程序包(vienna ab initio simulation program)[13],采用了Wang 和Perdew[14]發展的廣義的共軛梯度近似方法(generalized conjugate gradient approximation,GGA)描述電子交換-相關能,使用Vanderbilt 型超軟贗勢描述電子與離子間的相互作用[15]。在分子動力學模擬過程中,對每一構型都進行了電子結構的優化。模擬過程使用了正則系綜并采用Nosé 熱浴法來控制系統的溫度[16]。
較之傳統分子動力學模型,從頭算分子動力學模型計算量更大,目前計算體系多數在100 粒子左右。但是,由于從頭算動力學方法考慮了體系的電子結構,所得結果更可靠[17,18]。在55%Al-Zn 合金熔體結構的計算中,使用含有80個原子的立方晶胞模擬熔體結構(包含60個Al 原子和20個Zn 原子),溫度取為恒定的923K。并采用實驗密度,取變量點描述布里淵區。電子波函數向平面波展開,截斷能為250eV。用Verlet 算法求解離子的運動方程并取時間步長為3fs。平衡后,感興趣的物理量取自9ps 的統計平均值。同時,為驗證結果的正確性,也采用同樣的方法計算了Al 和Zn 熔體的結構參數,并與實驗結果進行了比較。
2 結構分析方法
在液態結構的分子動力學模擬研究中,各種物理量取系綜統計平均值。
2.1 結構因子
在計算中,取液態合金的偏結構因子為:
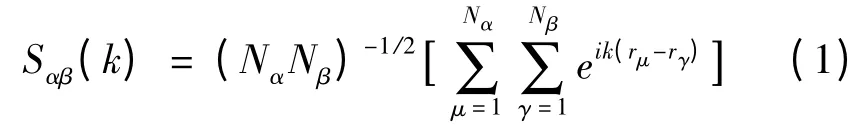
粒子的位置r 由從頭算分子動力學模擬得到;Nα是第α 種成分原子的數目;括號[…]代表對系綜和時間求平均。
2.2 雙體相關函數
在多元液體中以α 粒子為中心在r 處發現β 粒子的數密度為:
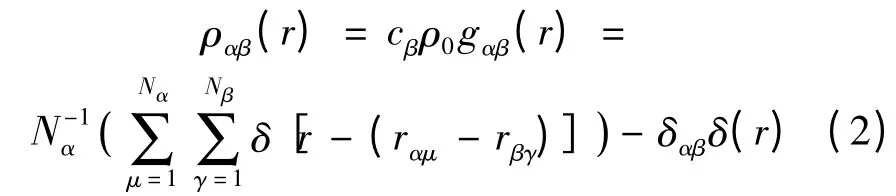
式中,Nα為第α 種成分的原子數。gαβ(r)即為偏雙體相關函數(partial pair correlation function)。
2.3 均方位移和速度自相關函數
分子動力學計算中原子不停地移動。以rαi(t)表示t 時刻第i個α 種粒子的位置,則粒子位移平方的平均值稱為均方位移(mean square displacement,MSD)為:
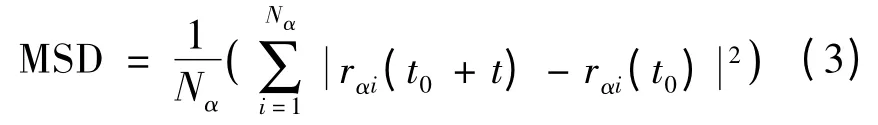
均方位移與原子擴散系數符合愛因斯坦擴散定律[19]:
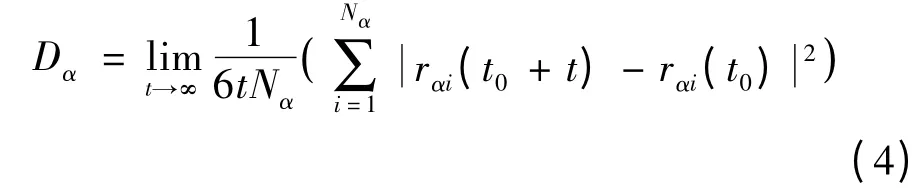
以Vα(t)表示t 時刻第α 種粒子的速度,則粒子的速度自相關函數(velocity autocorrelation function,VACF)定義為[20]:
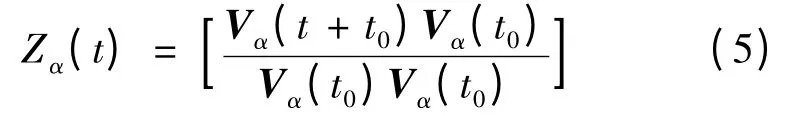
與之相應的光譜密度由下式給出[21]:
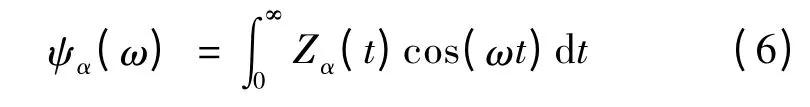
3 結果與討論
3.1 熔體結構因子計算值與實驗結果的比較
55%Al-Zn 合金熔體結構因子在實驗上采用X射線衍射測得。圖1 為55%Al-Zn 合金熔體結構因子的計算值與實驗結果的比較,從圖中可以看出這兩種方法得到的結果符合較好。由此可見,從頭算分子動力學方法對熔體結構的描述是合理的。
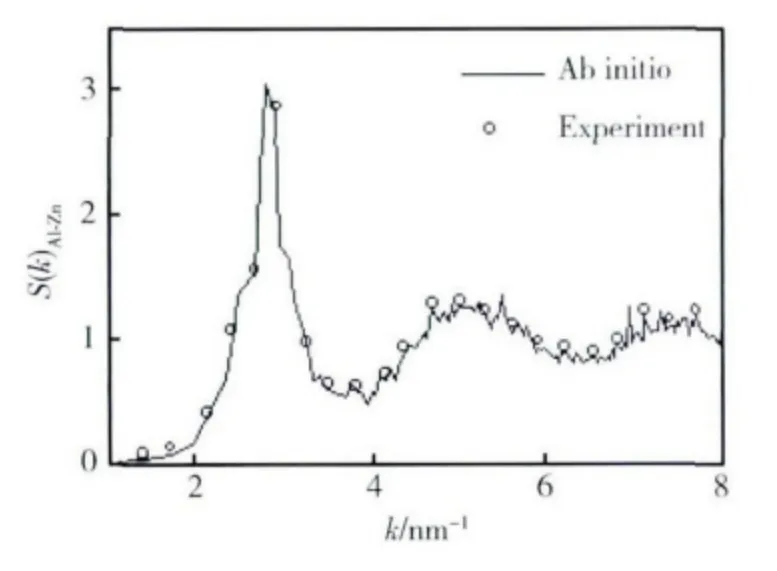
圖1 55%Al-Zn 合金熔體結構因子的計算值與實驗值Fig.1 The comparison of structure factor of 55% Al-Zn melt between experimental value and calculated value
3.2 55%Al-Zn 合金熔體的結構因子
計算得到的55%Al-Zn 合金熔體的三個偏結構因子分別見圖2,由圖2 可以看出,偏結構因子SAl-Zn(k)與SAl-Al(k)幾乎重合,表明Al 原子的分布在加入Zn 原子后變化不大。由圖2 可知,與偏結構因子SAl-Zn(k)相比,SZn-Zn(k)的第二峰變得非常平坦,表明Zn 原子基本上是隨機分布的。
3.3 55%Al-Zn 合金熔體的偏雙體分布函數
圖3 給出計算得到的55%Al-Zn 合金熔體的三個偏雙體分布函數,三個偏雙體分布函數第一峰的位置差別不大,在55%Al-Zn 合金熔體gZn-Zn(r)的第二峰消失,表明Zn-Zn 間的相關性發生明顯改變,而Al-Al 間的相關性變化不大,與55%Al-Zn 合金熔體的雙體分布函數幾乎重合。
由偏雙體分布函數可計算原子配位數:
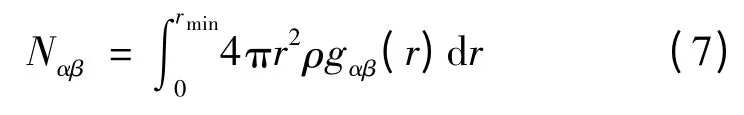
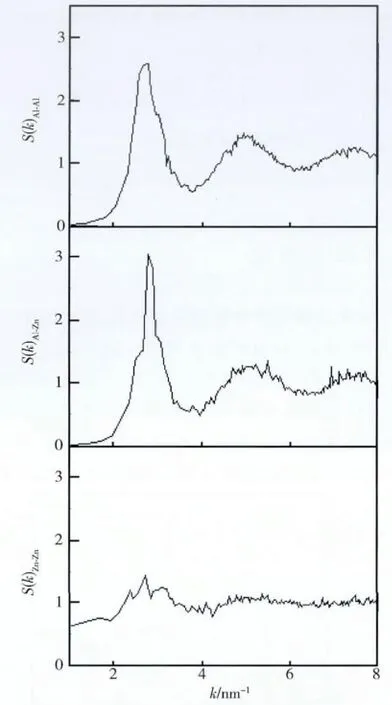
圖2 55%Al-Zn 合金熔體中結構因子Fig.2 The calculated structure factor of 55% Al-Zn alloy melt
計算中取截斷距離rmin為0.36nm,Al 熔體的配位數為10.10,Zn 熔體的配位數為10.61。Al-Zn 熔體中Al-Al,Al-Zn 和Zn-Zn 的配位數見表1。
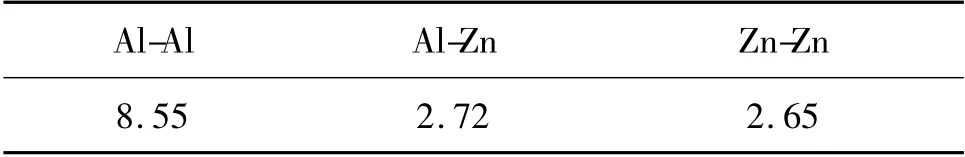
表1 Al-Zn 熔體中原子配位數Table 1 The atom coordination number of Al-Zn alloy melt
由表1 可以看出,Al-Zn 熔體中Al-Al 配位數與Al-Zn 配位數的比大約為3∶1,與熔體中原子含量的比值基本一致。這表明Zn 原子在Al-Zn 熔體中第一配位層內是按無規密堆分布的。再考慮到gZn-Zn(r)除存在第一峰外,其他峰幾乎觀察不到,可以認為Zn 原子在熔體中是均勻分布的。
3.4 原子擴散系數
原子在熔體中的擴散運動可通過研究原子的運動而得到。熔體中原子運動的均方位移見圖4。由式(5)得到的原子擴散系數見表2。Al 熔體的原子擴散系數缺乏實驗數據,本次研究的計算值與Protopapas 等人的理論預測值符合得較好[22]。Zn 熔體擴散系數的計算值與實驗值基本一致[23]。
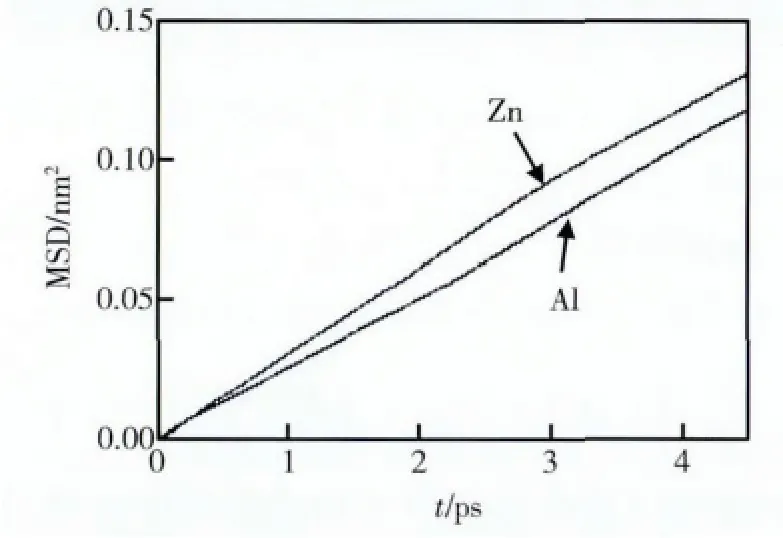
圖4 Al-Zn 熔體中原子的均方位移Fig.4 The mean square displacement of Al atom and Zn atom in Al-Zn alloy melt
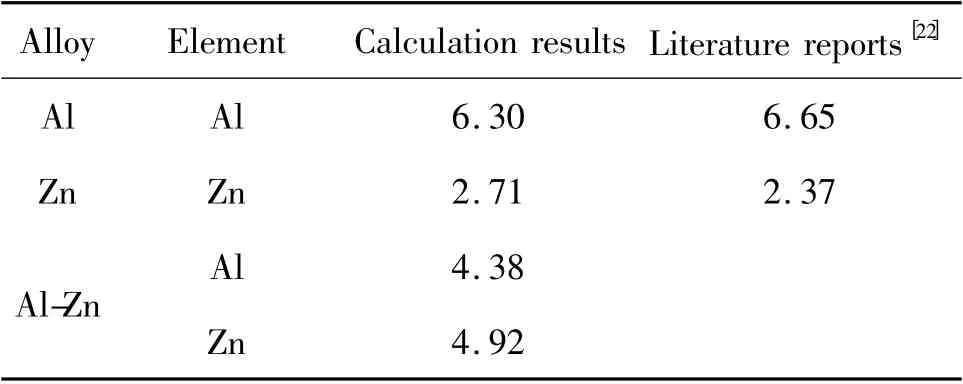
表2 Al,Zn 和55%Al-Zn 熔體中原子的擴散系數Table 2 The atom diffusion coefficient of Al,Zn,and Al-Zn alloy melt
3.5 原子速度自相關函數和光譜密度
原子在熔體中的擴散運動也可由速度自相關函數來表示。Al-Zn 熔體中原子的速度自相關函數見圖5。與Zn 原子相比Al 原子的速度自相關函數表現為明顯的第一谷,這表明周圍原子對Al 原子運動的阻礙作用比較強。這一方面與Al 原子的質量較小有關,另一方面是由于Zn 原子的原子半徑較小。
由速度自相關函數的傅里葉變換可得到原子運動的光譜密度函數,結果見圖6。由光譜密度函數可觀測原子運動的頻率,圖6 中顯示出原子Al 和Zn 在熔體中的“阻礙移動”,“阻礙移動”的意義為在熔體中原子的移動受到周圍原子的阻礙,結果類似于在一區間中來回移動。由圖6 可知,Al 原子光譜密度函數的峰值位于6THz 左右,遠大于Zn 原子的2THz,這也說明Al 原子周圍的第一近鄰Zn 原子形成的籠體對Al 原子運動的阻礙作用較強,同時也說明Al 和Zn 原子間有較強的相互作用。
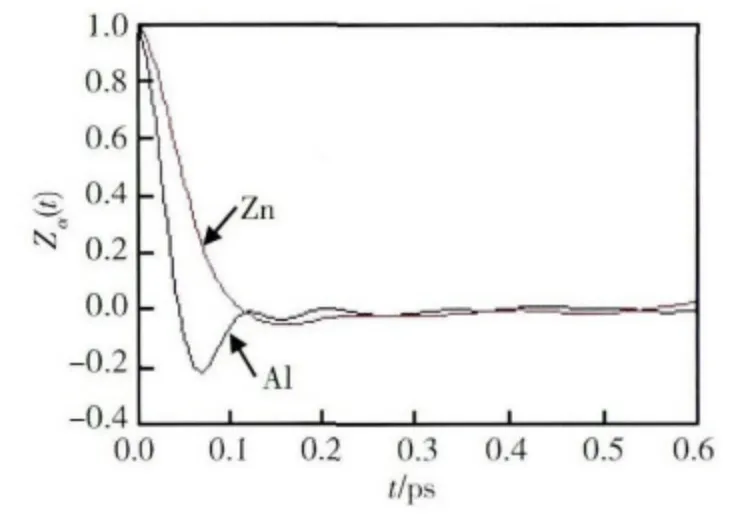
圖5 55%Al-Zn 熔體中原子速度自相關函數Fig.5 The velocity autocorrelation function of the atom in 55% Al-Zn alloy
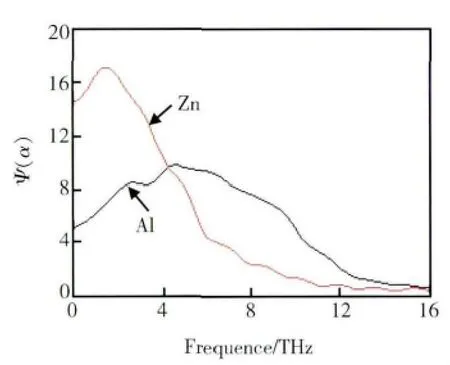
圖6 55%Al-Zn 熔體中原子光譜密度函數Fig.6 The spectral concentration function of the atom in 55% Al-Zn alloy
4 結論
(1)采用第一性原理分子動力學模擬方法研究高溫55%Al-Zn 合金熔體的結構和動力學性質,熔體結構中Al 原子的分布在加入Zn 原子后無明顯變化,而Zn 原子的分布是隨機均勻的。
(2)熔體中Al-Al 配位數與Al-Zn 配位數的比大約為3∶1,與熔體中原子含量的比值基本一致。
(3)熔體中Al 原子周圍的第一近鄰原子形成的籠體對Al 原子運動的阻礙作用較強。
[1]EGGELER G,AUER W,KAESCHE H. On the influence of silicon on the growth of the alloy layer during hot dip aluminizing[J]. Journal of Materials Science,1986,21(9):3348 -3350.
[2]SIMPSON T C. Accelerated corrosion test for aluminumzinc alloy coatings[J]. Corrosion,1993,49(7):550 -560.
[3]BARNARD N C,BROWN S G R. Modelling the relationship between microstructure of galfan-type coated steel and cut-edge corrosion resistance incorporating diffusion of multiple species[J]. Corrosion Science,2008,50(10):2846-2857.
[4]FELIU S,BARRANCO V. Study of degradation mechanisms of a protective lacquer film formulated with phosphating reagents applied on galvanised steel,galvanneal and galfan in exposure to UV/condensation test[J]. Surface &Coatings Technology,2004,182(2/3):251 -260.
[5]FELIU S,BARRANCO V. Effect of the incorporation of chromating reagents in an acrylic lacquer applied on galvanised steel,galvanneal and galfan during the UVCON test[J]. Surface & Coatings Technology,2004,182(2/3):318 -328.
[6]PUOMI P,FAGERHOLM H M,SOPANEN A. Parameters affecting long-term performance of painted galvanised steels[J]. Anti-Corrosion Methods and Materials,2001,48(3):160 -171.
[7]CONI N,GIPIELA M L,D'OLIECEIRA A S C M. Study of the mechanical properties of the hot dip galvanized steel and galvalume[J]. Journal of The Brazilian Society of Mechanical Sciences and Engineering,2009,31(4):319 -326.
[8]YANG D,CHEN J S,HAN Q. Effects of lanthanum addition on corrosion resistance of hot-dipped galvalume coating[J]. Journal of Rare Earths,2009,27(1):114 -118.
[9]LIVATYLI H,DUGGAL N,AHMETOGLU M A. Investigation of crack formation on the galvalume coating of roll formed roof panels[J]. Journal of Materials Processing Technology,2000,98(1):53 -61.
[10]GU T,QIN J,XU C,et al. An structural,bonding,and dynamical properties of liquid Fe-Si alloys:ab initiomolecular-dynamics simulation[J]. Phys Rev:B,2004,70(14):144204 -144210.
[11]GU T,BIAN X,QIN J,et al. Ab initio molecular dynamics simulations of liquid structure change with temperature for a GaSb alloy[J]. Phys Rev:B,2004,70(24):245214 -245220.
[12]GU T,BIAN X,QIN J,et al. Ab initio molecular-dynamics simulations of liquid GaSb and InSb[J]. Phys Rev:B,2005,71(10):104206 -104213.
[13]KRESSE G,FURTHMüLLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Comput Mater Sci,1996,6(1):15 -50.
[14]WANG Y,PERDEW J P. Correlation hole of the spin-polarized electron gas,with exact small-wave-vector and high-density scaling[J]. Phys Rev:B,1991,44(24):13298 -13307.
[15]VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Phys Rev:B,1990,41(11):7892 -7895.
[16]NOSé S. A unified formulation of the constant temperature molecular dynamics methods[J]. Chem Phys,1984,81(1):511 -519.
[17]CAR R,PARRINLLO M. Structural,dynamical,and electronic properties of amorphous silicon:an ab initio molecular-dynamics study[J].Phys Rev Lett,1988,60(3):204 -207.
[18]SENDA Y,SHIMOJO F,HOSHINO K. The ionic structure and the electronic states of liquid Li-Pb alloys obtained from ab initiomolecular dynamics simulations[J]. J Phys:Condens Matter,2000,12(28):6101 -6112.
[19]MARK A,DANE M,HOYT J,et al. Embedded-atommethod study of structural,thermodynamic,and atomictransport properties of liquid Ni-Al alloys[J]. Phys Rev:B,1999,59(22):14271 -14281.
[20]JEROME J E,WILLIAM W W. Molecular-dynamics calculations of the velocity-autocorrelation function:methods,hard-disk results[J]. Phys Rev:A,1982,26(3):1648-1675.
[21]谷廷坤. 合金熔體局域結構的實驗及從頭算研究[D].山東:山東大學 材料科學與工程學院,2005.
[22]PROTOPAPAS P,PARLEE N A D. Theory of transport in liquid metals :Ⅲ:calculation of shear viscosity coefficients of binary alloys[J]. Chemical Physics,1975,11(1):201 -215.
[23]NACHTRIEB N H,FRAGA E,WAHL C. Self-diffusion of liquid zinc[J]. J Phys Chem,1963,67(11):2353 -2355.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
