La摻雜對Cr2O3催化劑氣相氟化四氯乙烯反應性能的影響
2014-10-13 07:57:52范鏡蓮程永香趙洋王芳羅孟飛王月娟
化工進展 2014年1期
關鍵詞:催化劑
范鏡蓮,程永香,趙洋,王芳,羅孟飛,王月娟
(浙江師范大學物理化學研究所,浙江省固體表面反應化學重點實驗室,浙江 金華 321004)
五氟乙烷(簡稱HFC-125)的消耗臭氧潛能值(ODP)為0,全球變暖潛能值(GWP)為2800,是R404A、R407C、R410、R402A、R507等環保型混合制冷劑的主要組分,這些環保型混合制冷劑可
以替代現有的二氟一氯甲烷(HCFC-22)制冷劑。HFC-125還廣泛用作發泡劑、溶劑、噴射劑和干蝕刻劑[1]。
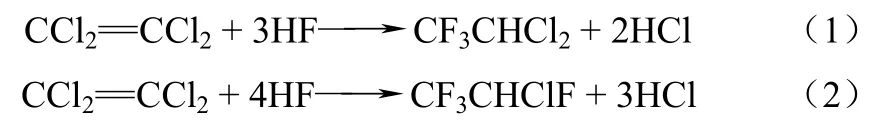
氟化含氯烯烴是合成氫氟烴(HFCs)的主要合成路線,如以三氯乙烯為原料合成 HFC-134a(CF3CH2F)[2];四氯乙烯為原料合成HFC-125[3-4]。工業上通常采用液相和氣相氟化兩種氟化路線,前者由于對設備腐蝕性大、環境污染嚴重逐漸被淘汰[4],因此目前普遍采用的是氣相氟化路線。四氯乙烯路線分兩個步驟來實現:第一步是氟化四氯乙烯(簡稱PCE)制備HCFC-123(2,2-二氯-1,1,1-三氟乙烷)和HCFC-124(2-氯-1,1,1,2-四氟乙烷),反應式如式(1)、式(2)。
第二步由HCFC-123或HCFC-124進一步氟化制備HFC-125,反應如式(3)、式(4)。
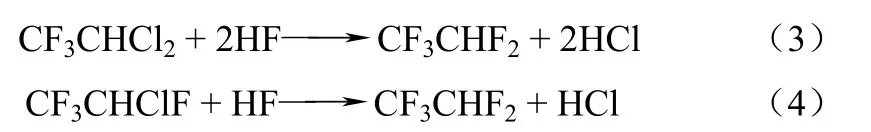
氣相氟化普遍采用 Cr基催化劑,有負載型Cr/AlF3[4]、Cr/Al2O3[5]、Cr/MgO 和 Cr/MgF2[6]等催化劑;非負載的CrOx[7]催化劑;摻雜的Cr2O3[8]催化劑。雖然這些催化劑已經廣泛用于HFCs的工業化合成,但是對催化劑的活性物種及其反應機理尚沒有統一的認識。很多研究認為催化劑表面CrOF是氣相氟化反應的活性中心,如Lee等[6]研究發現,Cr/MgF2催化劑活性要比純CrOx催化劑活性高,他們認為催化劑的活性中心為 CrOxFy,而非 CrF3。Adamczyk等[9]用(NH4)3CrF6和Cr2O3在高溫下制備了一系列的CrOxFy催化劑,并用XRD和XANES對催化劑進行了表征,實驗表明含有CrOxFy物種催化劑表現出比純Cr2O3和CrF3更高的活性,是該催化劑的活性中心。Rao等[10]制備了 Cr2O3、Cr2O3/Al2O3(浸漬法)及 Cr2O3/Al2O3(共沉淀法)催化劑,用 XPS技術對 Cr物種進行了表征,認為Cr(OH)F2為活性物種。另外,有人認為催化劑中高價態的 Cr有利于氟化反應[11],還有人認為催化劑的活性依賴于催化劑表面的酸性位[12-13]。總而言之,目前關于 Cr基催化劑的活性物種還沒有一致的認識,還需要進一步的考察和研究。
先前作者[14]報道了ZnO/Al2O3催化劑對氣相氟化四氯丙烯反應性能的催化性能,考察了載體Al2O3的焙燒溫度對催化劑性能的影響,發現Al2O3載體經過1100 ℃焙燒制得的ZnF2/Al2O3催化劑催化性能最高,當反應溫度為300 ℃時,四氯乙烯的轉化率為 45.7%,HCFC-123(2,2-二氯-1,1,1-三氟乙烷)和HCFC-124(2-氯-1,1,1,2-四氟乙烷)的總選擇性為 48.2%。然而,催化劑的轉化率和選擇性較低,無法滿足工業化需要。
本文以 Cr2O3為基礎,通過向其中添加不同含量的La助劑,研究不同含量的La助劑摻雜對Cr2O3催化劑氟化四氯乙烯性能的影響及其表面酸性、CrOxFy物種對催化劑活性的影響。
1 實驗部分
1.1 實驗試劑
九水硝酸鉻,AR,國藥集團化學試劑有限公司;六水硝酸鑭,AR,國藥集團化學試劑有限公司;濃氨水,AR,杭州余杭利人醫藥化工有限公司;四氯乙烯,純度>99%,浙江三美化工股份有限公司提供;無水HF,純度>99.9%,浙江三美化工股份有限公司;蒸餾水,自制。
1.2 催化劑制備
La2O3-Cr2O3催化劑采用沉淀法制備。稱取一定量的 Cr(NO3)3·9H2O 和 La(NO3)3·6H2O 配成混合溶液,然后用沉淀劑氨水沉淀,分離得到的沉淀物經烘干(120 ℃),在500 ℃氮氣氣氛下焙燒4 h,即制得xLa2O3-Cr2O3催化劑前體,x為催化劑中La摩爾分數,分別為0、1%、5%和10%。將La2O3-Cr2O3催化劑前體裝入自制的反應裝置(內徑為10 mm的不銹鋼反應管),通入N2-HF混合氣體(HF與 N2的摩爾比為4,總流量為50 mL/min),升溫至260 ℃保持1 h,再升溫至400 ℃保持3 h,得到氟化后的催化劑,催化劑記為 xLaF3-Cr2O3。Cr2O3(F)、1LaF3-Cr2O3、5LaF3-Cr2O3、10LaF3-Cr2O3催化劑的比表面積分別為53 m2/g、49 m2/g、47 m2/g和42 m2/g。
LaF3采用LaF3-Cr2O3催化劑相同的方法制備。氟化預處理后的樣品記為LaF3,LaF3的比表面積為12 m2/ g。
CrF3的制備:稱取一定量的Cr(NO3)3·9H2O配成溶液,然后用沉淀劑氨水沉淀,分離得到的沉淀物經烘干(120 ℃),即制得 Cr(OH)3前體。將Cr(OH)3前體裝入自制的反應裝置(內徑為10 mm的不銹鋼反應管),經200 ℃干燥2 h后,在N2-HF混合氣體(HF與 N2的摩爾比為 4,總流量為 50 mL/min)下升溫至 300 ℃氟化 10 h得到無定形CrF3,其比表面積為68 m2/g。
1.3 催化劑表征
X射線衍射(XRD)分析采用荷蘭Philips公司生產的PW3040/60型全自動X射線衍射儀。Cu Kα射線,管電壓40 kV,管電流40 mA,掃描速度0.15°/s,掃描范圍2θ 為10°~80°。所有XRD測試均在靜態空氣氣氛下進行。
催化劑的比表面積測定采用美國Quantachrome Autosorb-1型N2物理吸附儀測定。所有測試樣品在300℃真空預處理4h,與液氮溫度下進行實驗。采用BET公式計算樣品的比表面積。
催化劑的表面酸性通過氨氣程序升溫脫附(NH3-TPD)實驗來測試。實驗在自制的裝置上進行,將100 mg催化劑裝入直徑為6 mm的石英管反應器中,于350 ℃氮氣氣氛下干燥1.5 h,除去催化劑中的水分,然后降溫至50 ℃吸附NH330 min,再于100 ℃下氮氣吹掃90 min以除去殘留的和物理吸附的 NH3。然后以 10 ℃/min的速率升溫至600 ℃進行NH3脫附,脫附的NH3通過TCD檢測器檢測。
X射線光電子能譜是在VGESCALAB MK-2型能譜儀上測定,AlKα射線源(1486.6 eV),加速電壓20 kV,樣品以C1s=284.6 eV為基準進行結合能校正。
1.4 催化劑活性評價
在自制的內徑為 10 mm的不銹鋼管固定床反應器中裝入3 mL催化劑,熱電偶放在催化劑床層的中間監控反應溫度。控制 HF氣體的流量為 20 mL/min,反應物中HF與四氯乙烯摩爾比為13∶1。反應物和產物用氣相色譜(島津GC-2014)進行在線分析,使用氫離子火焰檢測器(FID)和GS-GASPRO毛細管柱(60 m×0.32 mm)。
2 結果與討論
2.1 催化劑的物相分析
圖 1為 La2O3-Cr2O3和 LaF3-Cr2O3催化劑的XRD圖譜。對于La2O3-Cr2O3催化劑[圖1(a)],Cr2O3和1La2O3-Cr2O3樣品只觀察到Cr2O3的特征衍射峰,表明Cr物種以Cr2O3的形式存在,沒有檢查到La的相關衍射峰,表明低含量的 La以無定形的形式存在,或含量太低XRD檢測不到。當La含量達到5%時,除了Cr2O3的特征衍射峰,還觀察到LaCrO4和LaCrO3的特征衍射峰,表明La含量的增加,有利于形成La-Cr復合氧化物。從La2O3-Cr2O3催化劑前體經氟化預處理后的 LaF3-Cr2O3催化劑[圖 1(b)]的XRD圖可以看出,催化劑中Cr2O3的特征衍射峰依然存在,這說明在氟化預處理的過程中晶相Cr2O3很難被氟化成 CrF3,這與文獻中報道是一致的[10,15]。文獻[16]中指出,由 Cr2O3和 HF反應制CrF3,該反應具有較小負值的吉普斯自由能(ΔrG=?68 kJ/mol)。這同樣也說明該反應也較難進行或者說該反應需要較長的時間才能有較大程度的反應。預氟化后的催化劑除了有Cr2O3特征衍射峰外,同時還出現了LaF3的特征衍射峰,并且這些峰隨著 La含量的增加變得越來越強,然而,LaCrO4和LaCrO3的特征衍射峰卻消失了,表明LaCrO4和LaCrO3完全轉化成LaF3和CrF3(或Cr2O3)。由于La2O3容易氟化生成LaF3[17],因此1LaF3-Cr2O3催化劑中沒有觀察到 LaF3的特征衍射峰可能與助劑的添加量較少,生成的 LaF3均勻分散在 Cr2O3表面有關。
圖2是LaF3-Cr2O3催化劑的Raman圖譜。對于LaF3-Cr2O3催化劑,從圖2中可以看出,在302 cm?1、350 cm?1、550 cm?1和 611 cm?1處出現 4 個 Raman振動峰,這4個峰都歸屬為晶相Cr2O3的Raman振動峰[18]。這說明通過氟化預處理后Cr2O3依然存在,這與XRD結果是一致的,進一步說明晶相的Cr2O3很難被氟化。

圖1 La2O3-Cr2O3和LaF3-Cr2O3催化劑的XRD圖譜
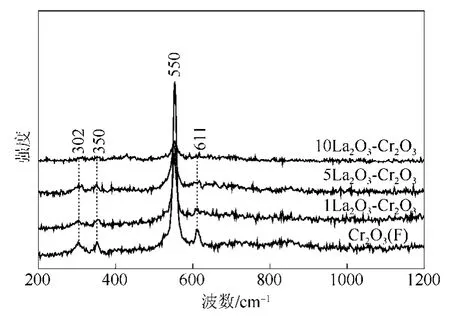
圖2 LaF3-Cr2O3催化劑的Raman圖譜
2.2 催化劑的NH3-TPD表征
圖 3是 Cr2O3(F)和 LaF3-Cr2O3催化劑的 NH3-TPD圖,為了對比,圖3中也給出了LaF3和CrF3的NH3-TPD圖。從圖3中可以看出,Cr2O3(F)催化劑在150~550 ℃之間出現分布范圍較寬的NH3脫附峰,表明酸性位分布較廣,包含弱酸性位和強酸性位。1LaF3-Cr2O3催化劑的NH3脫附峰與Cr2O3(F)催化劑基本相同。仔細觀察可以發現,1LaF3-Cr2O3催化劑低于200 ℃沒有發現NH3脫附,表明部分弱酸中心消失。然而,當La添加量增加到5%和10%時,在450 ℃附近出現一個很強的NH3脫附峰,低溫區(170~300 ℃)的 NH3脫附峰消失,催化劑表面酸性位分布變窄,峰面積明顯增加。這表明隨著 La含量的增加,LaF3-Cr2O3催化劑弱酸中心減少,催化劑表面酸性位的性質發生改變,催化劑表面只有強酸中心。通過對 NH3-TPD過程流出NH3的定量計算,Cr2O3(F)、1LaF3-Cr2O3、5LaF3-Cr2O3、10LaF3-Cr2O3、LaF3和CrF3樣品表面脫附的NH3(表面酸量)分別為 83 μmol/g、58 μmol/g、100 μmol/g、232 μmol/g、78 μmol/g 和 346 μmol/g。根據催化劑的表面積和表面酸量可計算得到催化劑的表面酸密度。Cr2O3(F)、1LaF3-Cr2O3、5La F3-Cr2O3和10La F3-Cr2O3的表面酸密度分別為1.57 μmol/m2、1.18 μmol/m2、2.13 μmol/m2和 5.52 μmol/m2;LaF3和 CrF3的表面酸密度分別為 6.50 μmol/m2和 5.09μmol/m2。可見少量La的添加使得表面酸密度減少;La添加量達到5%后,由于生成較多的LaF3和CrF3,催化劑表面酸量明顯增加。
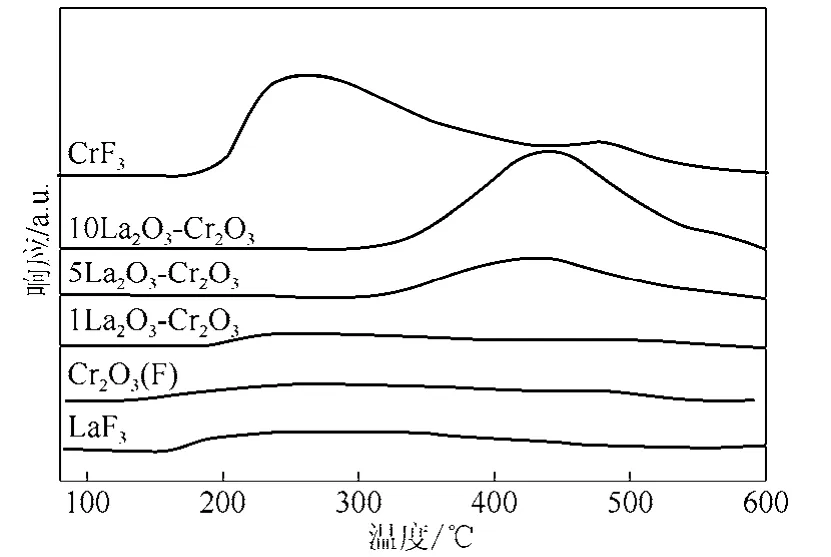
圖3 Cr2O3(F)和LaF3-Cr2O3催化劑的NH3-TPD圖
2.3 催化劑的XPS表征
圖4是LaF3-Cr2O3催化劑的Cr2p XPS圖譜。由圖4可以看到,在結合能為575.5 eV和577.5 eV分別出現了兩個峰值,這些峰值的出現歸功于Cr3+的2p3/2電子,根據文獻[11,19],峰值為575.5 eV和577.5 eV的兩個峰分別歸屬為Cr2O3和CrOxFy,可見LaF3-Cr2O3催化劑表面存在Cr2O3和CrOxFy。表 1是 LaF3-Cr2O3催化劑表面 CrOxFy相對含量和Cr2p2/3的結合能。催化劑表面CrOxFy含量由圖4中CrOxFy的峰面積與Cr2O3和CrOxFy兩個峰的總面積相比計算得到。從表1中可以看出,隨著LaF3-Cr2O3催化劑中 La含量的增加,CrOxFy物種的結合能往更高結合能方向偏移,這可能是因為 La的含量與Cr2O3的相互作用造成的。1LaF3-Cr2O3催化劑的表面 CrOxFy含量(35.7%)低于 Cr2O3(F)催化劑表面CrOxFy含量(36.5%),然而進一步增加La的含量,催化劑表面CrOxFy含量增加,這表明La的添加量影響LaF3-Cr2O3表面CrOxFy的含量。
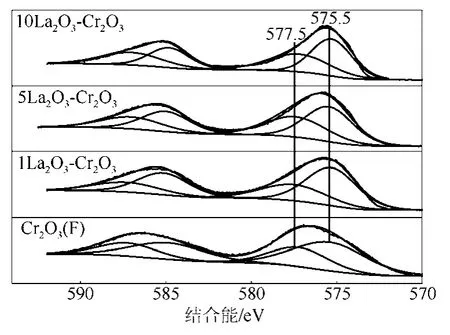
圖4 LaF3-Cr2O3催化劑的Cr2p XPS圖譜
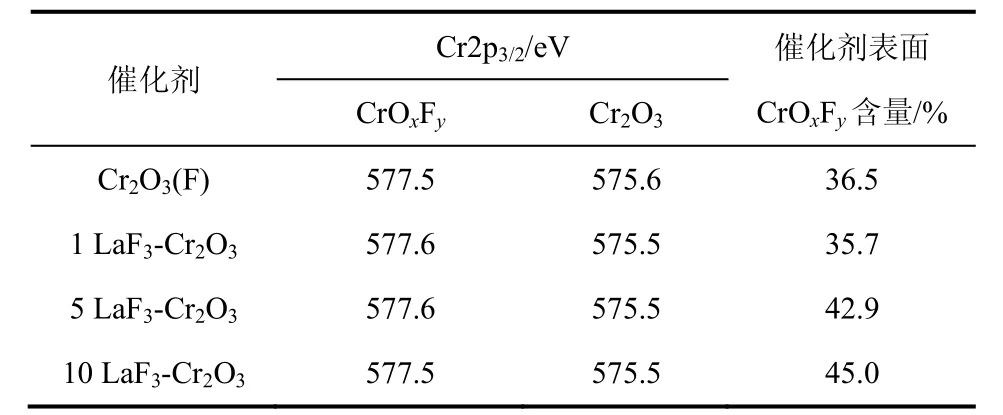
表1 LaF3-Cr2O3催化劑表面CrOxFy含量和Cr 2p2/3的結合能
2.4 LaF3-Cr2O3催化劑催化劑的四氯乙烯氟化反應性能
表2列出了LaF3-Cr2O3催化劑的四氯乙烯氟化反應的轉化率、選擇性和反應比速率(數據取自反應10 h)。從表2中可以看出,隨著La含量的增加,PCE的轉化率逐漸增加,由Cr2O3催化劑的81.1%到10LaF3-Cr2O3催化劑的98.7%,催化劑的比速率總體上隨著催化劑表面酸密度的增加而增加。然而,HCFC-123、HCFC-124和HFC-125的總選擇性卻先增加后減少。其中1LaF3-Cr2O3催化劑上HCFC-123、HCFC-124和HFC-125的總選擇性最高(90.1%)。然而隨著 La含量的繼續增加,HCFC-123、HCFC-124和HFC-125的總選擇性快速下降,當La含量為10%(摩爾分數)時,10LaF3-Cr2O3催化劑上HCFC-123、HCFC-124和HFC-125的總選擇性僅為 34.7%。同時,隨著 La含量的增加,副產物HCFC-133a的選擇性逐漸增加,當La含量為10%(摩爾分數)時,HCFC-133a的選擇性高達31.4%。
將催化劑表面酸密度與HCFC-123、HCFC-124和HFC-125選擇性總和作圖,得到催化劑表面酸量與產物總選擇的關系圖(圖5)。從圖5中可以看出,隨著酸量的增加,HCFC-123 、HCFC-124和HFC-125的總選擇性逐漸減少。這說明催化劑表面酸中心的增加不利于 HCFC-123、HCFC-124和HFC-125產物的生成。從圖5還可以看出,1LaF3-Cr2O3催化劑表面酸密度低于 Cr2O3(F),有利于HCFC-123、HCFC-124和 HFC-125的生成;當La添加量達到 5%后,催化劑表面酸量明顯增加,提高了反應比速率,但不利于HCFC-123、 HCFC-124和 HFC-125的生成。也發現 HCFC-133a和 HFC-134a等副產物隨著催化劑酸量的增加而增加,其中HCFC-133a的量增加尤其明顯,當催化劑添加了5%的La時,HCFC-133a的選擇性為16.4%,當催化劑添加了 10%的 La時,HCFC-133a的選擇性為31.4%,這說明表面酸密度與目標產物(HCFC-123、HCFC-124和 HFC-125)總選擇性存在對應關系,即催化劑表面相對酸密度的增加不利于HCFC-123、HCFC-124和HFC-125的生成。

表2 LaF3-Cr2O3上PCE的轉化率,HCFC-123、HCFC- 124、HFC-125的選擇性和反應的比速率(數據取自反應10 h)
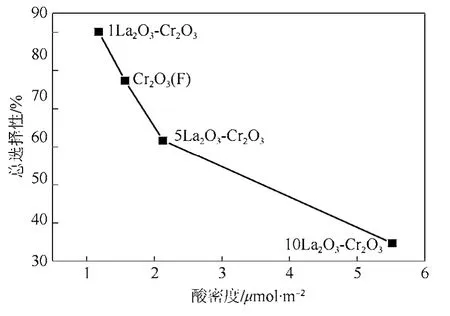
圖5 催化劑表面酸密度與產物總選擇(HCFC-123 +HCFC-124+ HFC-125)的關系圖
催化劑表面酸性增加往往有利于歧化反應的發生[13,20-21],Hess 等[12]通過比較 β-AlF3和 AlF2(OH)催化劑的 CHClF2歧化反應,發現表面酸中心有利于歧化反應的發生。然而表面酸性也有利于裂解反應的發生[22-25]裂解反應不僅需要酸性活性中心,而且能斷裂C—C鍵的強酸中心[26]。由于催化劑表面酸性增加有利于歧化和裂解反應,因此過多的 La使得催化劑表面酸性增加從而導致副反應的發生,使得有效產物(HCFC-123、HCFC-124和HFC-125)的選擇性下降。
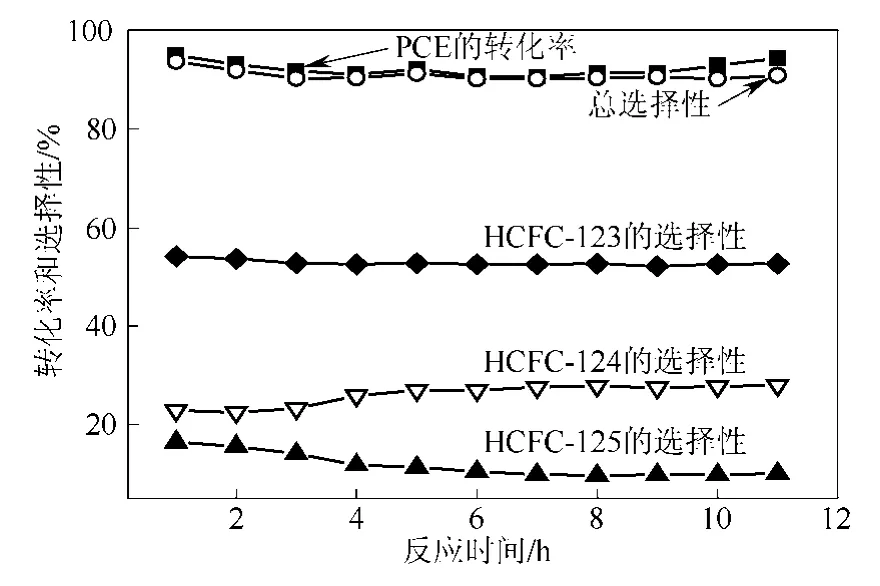
圖6 1LaF3-Cr2O3催化劑上氟化PCE的反應穩定性(反應溫度為300 ℃)
圖6是在300 ℃下1LaF3-Cr2O3催化劑上氟化PCE反應的穩定性測試結果。從圖6中可以看出,PCE的轉化率和HCFC-123、HCFC-124和HFC-125的總選擇性在反應開始前3 h有輕微的下降,PCE的轉化率由 94.9%降到 91.0 %,HCFC-123、HCFC-124和 HFC-125的總選擇性由 93.6%降到90.1%。在此之后,PCE和HCFC-123、HCFC-124和HFC-125的總選擇性保持穩定,這說明該催化劑具有良好的穩定性。
3 結 論
LaF3-Cr2O3催化劑具有較好的催化氟化四氯乙烯反應性能,隨著La摻雜含量的增加,LaF3-Cr2O3催化劑上 PCE的轉化率不斷升高,而有效產物(HCFC-123 + HCFC-124 + HFC-125)總選擇性先升后降。當La摻雜為1%時,1LaF3-Cr2O3催化劑性能最佳,PCE的轉化率和有效產物總選擇性分別為92.8%和90.1%。結果表明,催化劑表面強酸中心不利于有效產物的生成。
[1]劉坤峰,楊會娥,張文慶,等. 氣相法合成五氟乙烷(HFC-125)的研究進展[J]. 有機氟工業,2009(1):55-58.
[2]Quan H D,Li Z,Zhao Z X,et al. Preparation of 1,1,1,2-tetrafluoroethane (HFC-134a) by vapor-phase catalytic fluorination[J]. Appl. Catal. B,1996,8:209-215.
[3]Yoshimura T,Homoto Y,Yamada Y,et al. Process for producing pentafluoroethane and tetrafluoroethane:US,6011185[P].2000-01-04.
[4]Cheminal B,Lacroix E,Lants A,et al. Process for fluorination of perchloroethylene or of pertachloroethane:US,5932776[P].1999-08-03.
[5]Cho D H,Kim Y G,Chung M J,et al. Preparation and characterization of magnesia-supported chromium catalysts for the fuorination of 1,1,1-triuoro-2-chloroethane (HCFC-133a)[J]. Appl.Catal. B,1998,18:251-261.
[6]Lee H,Jeong H D,Chung Y S,et al. Fluorination of CF3CH2Cl over Cr-Mg fluoride catalyst:The effect of temperature on the catalyst deactivation[J]. J. Catal.,1997,169:307-316.
[7]Brunet S,Requieme B,Colnay E,et al. Catalytic gas-phase fluorination of 1,1,l-trifluoro-2-chloroethane over chromium(Ⅲ)oxide:Preparation of hydrofluoroalkanes[J]. Appl. Catal. B,1995,5:305-317.
[8]Krishna Murthy J,Gross U,Rüdiger S,et al. Synthesis and characterization of chromium(Ⅲ)-doped magnesium fluoride catalysts[J]. Appl. Catal. A,2005,282:85-91.
[9]Adamczyk B,Boese O,Weiher N,et al. Fluorine modified chromium oxide and its impact on heterogeneously catalyzed fluorination reactions[J]. J. Fluorine Chem.,2000,101:239-246.
[10]Rao J M,Sivaprasad A,Narsaiah B,et al. A comparative study of bulk and supported chromia catalysts for the fluorination of trichloroethylene[J]. J. Catal.,1999,184:105-111.
[11]Quan H D,Tamura M,Matsukawa Y,et al. Investiganiton into chromia-based catalyst and its application in preparing difluoromethane[J]. J. Mol. Catal. A:Chem.,2004,219:79-85.
[12]Hess A,Kemnitz E. Characterization of catalytically active sites on aluminum oxides,hydroxyfluorides,and fluorides in correlation with their catalytic Behavior[J]. J. Catal.,1994,149(2):449-457.
[13]Brunet S,Boussand B,Barrault J,et al. Catalytic fluorination over chromium oxides. Preparation of hydrofluorocarbons[J]. Stud. Surf.Sci. Catal.,1996,101:379-385.
[14]程永香,謝遵運,羅孟飛,等. 氣相氟化四氯丙烯的 ZnF2/Al2O3催化劑研究[J]. 化工進展,2012,31(11):2483-2487.
[15]Xing L Q,Bi Q Y,Meng F L,et al. In situ Raman spectroscopy studies on chromium oxide catalyst in an anhydrous hydrogen fluoride atmosphere[J]. J. Raman Spectrosc.,2011,42:1095-1099.
[16]Bonniface D W,Scott J D,Watson M J,et al. Halogen exchange reactions for CFC alternatives:The behaviour of fluorine-18 labeled hydrogen fluoride towards prefluorianted chromia containing nickl(Ⅱ) orzinc (Ⅱ)[J]. Green Chem.,1999,1(1):9-11.
[17]Bi Q Y,Lu J Q,Meng F L,et al. Effect of calcination temperature on La-modified Al2O3catalysts for vapor phase hydrofluorination of acetylene to vinyl fluoride[J]. Chin. J. Chem. Phys.,2010,23:89-94.
[18]Barshilia H C,Rajam K S. Growth and characterization of chromium oxide coating prepared by pulsed-direct current reactive unbalanced magnetron sputtering[J]. Appl. Surf. Sci.,2008,255:2925-2931.
[19]Loustaunau A,Fayolle-Romelaer R,Celerier S,et al. Catalytic fluorination of various chlorinated hydrocarbons by HF and a chromium based catalyst:Effect of the presence of zinc[J]. Catal.Lett.,2010,138:215-223.
[20]Bell T N,Kirszensztejn P,Czajka B. Catalytic conversion of CC12F2on a γ-A12O3catalyst[J]. Catal. Lett.,1995,30:305-312.
[21]Hess A,Kemnitz E. Surface acidity and catalytic behavior of modified zirconium and titanium dioxides[J]. Appl. Catal. A:General,1997,149:373-389.
[22]Yusaku T,Tatsumi I. Catalytic decomposition of CFCs[J]. Catalysis Surveys from Japan,1998,2:165-173.
[23]Yusaku T,Maiko N,Rie M,et al. Decomposition of chloro ?uorocarbons over metal phosphate catalysts Part Ⅰ. Decomposition of CCl2F2over metal phosphate catalysts[J]. Phys. Chem. Chem.Phys.,1999,1:2367-2372.
[24]Masahiro T,Miki N,Yasushi F,et al. Decomposition of chlorofluorocarbons in the presence of water over zeolite catalyst[J].Appl. Catal. B:Environmental,1996,9:167-177.
[25]潘思忠,楊雪敏,高滋. 相同硅鋁比的ZSM-5、ZSM-35和Md沸石的表面酸性與催化性能[J]. 催化學報,1988,9(2):158-164.
[26]Corna A. Application of zeolites in fluid catalytic cracking and related processes[J]. Stud. Surf. Sci. Catal.,1989,49:49-67.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
